CASTEPを用いたELNES/XANESの第一原理計算
東京大学・生産技術研究所・溝口照康 Ver.20180815
以前に作成したマニュアル(2014年度版)からMaterials
Studioの仕様が変更されたため改訂しました.以前のバージョンはこちらです.
本ページでは第一原理擬ポテンシャル法であるCASTEPコードを用いたELNES/XANESの第一原理計算法を説明します.ダッソーシステムのMaterials
StudioというGUIを用いた手法について説明します.本稿では例として酸化マグネシウム(MgO)のO-K端を計算します.CASTEPでは.cell,.param,.castepの拡張子をもったファイル使いますが,拡張子前の名前について,本稿ではcase.castepとしております.
1.Materials Studio上でモデルを作製する.
まずMgOのモデルを作成します.Materials ProjectやMat naviなどのデータベースからも取得できますし,空間群と原子座標からも作成することができます.ここではMaterials Studioのメニュー File-> Importを行い,「Structures」のショートカットに行き,「Metal-oxides」の中のMgO.msiを呼び出します.
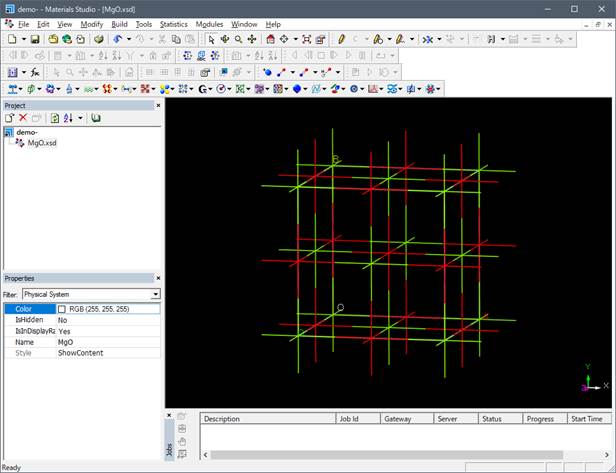
上図では酸素は赤色,Mgは緑色となっています.この構造は岩塩型構造の単位格子(unit
cell)ですので基本単位格子(primitive cell)ににして計算コスト削減します.
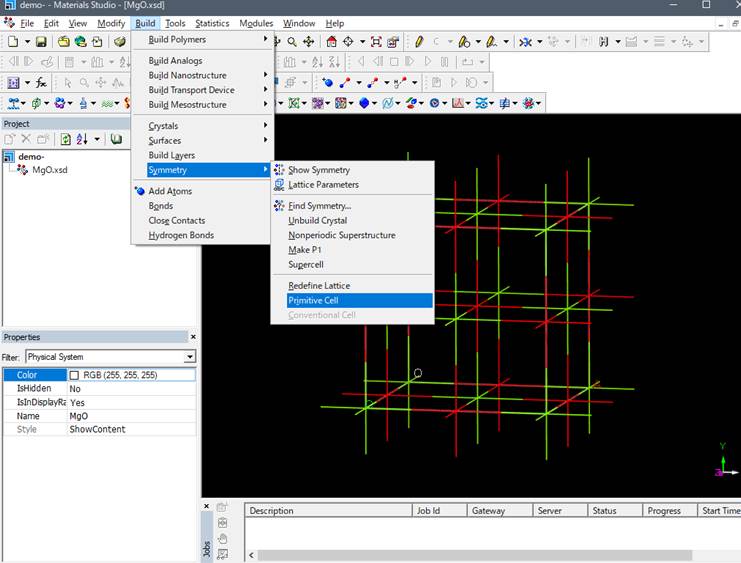
Mg 1 O
1の2原子で構成されたMgOのprimitive cellができます.基底状態計算(バンド構造や状態密度など)はprimitive cellを用いてk点を増やした計算を行います.しかし,ELNES/XANES計算で必須となる内殻空孔(Core-hole)効果をprimitive cellでは正確に計算できません.
内殻空孔は電子状態に影響を及ぼします.バンド計算では計算するセルを拡張する(周期的境界条件:Periodic boundary condition)ため,隣のセルの中にある内殻空孔を導入した原子 (Excited atom)間に相互作用が発生します(下図).
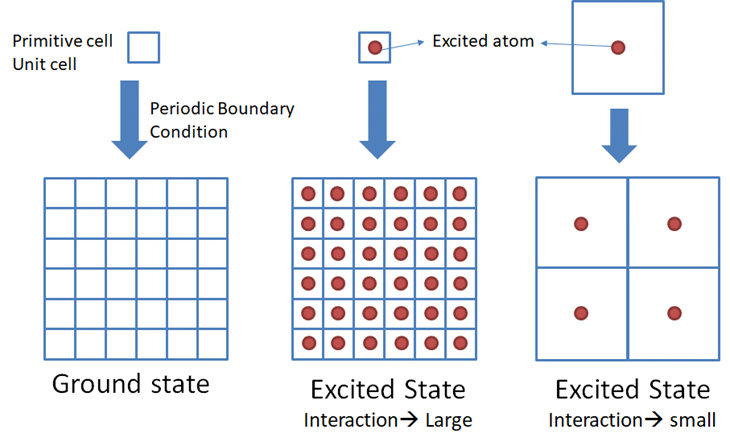
そこで,primitive cellやunit cellのような小さなセルではなく,それらを拡張したモデル(スーパーセル)を用い,内殻空孔を導入した原子間の相互作用を小さくします.これまでの研究で内殻空孔を導入した原子間を1nm(10Å)離す必要があることが分かっています.ここでは簡単のためにMgOのprimitive cellを下のように2x2x2倍に拡張したスーパーセルを使用します.
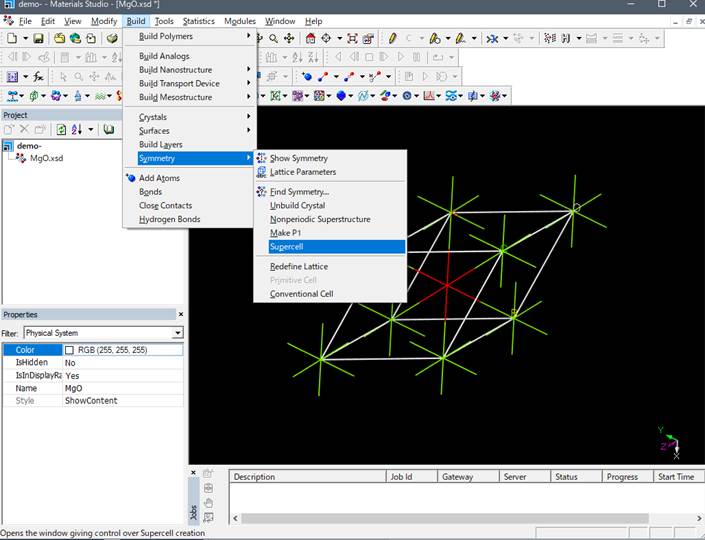
例えば2×2×2倍することで以下のような16原子のスーパーセルが作製されます.
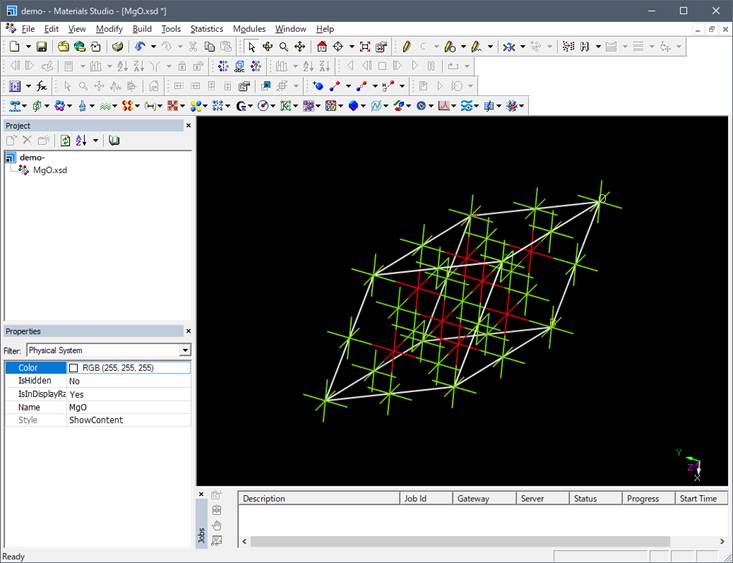
Materials Studioではsupercellにした時点で結晶の対称性はP1(対称性なし)に落ちます.
本来は約1nmのスーパーセルを用いる必要があるため,MgOの場合はprimitive cellを4×4×4倍した以下のような128原子のスーパーセルを用いるべきです.
(128原子の計算時間はSCF計算のみであれば4コアのノートパソコンで数十分,ELNES/XANES計算も行うと数時間です).
2.内殻空孔(Core-hole)を導入する
今回は酸素のK端を計算するので,酸素原子を選びます.どの酸素を選んでも問題ありません.「Line」表示だと分かりにくいので,右クリックのDisplay styleで「Ball and stick」表示にし,酸素を1つ選びます.ここではセル中の左下の酸素(黄色)を選択してます.
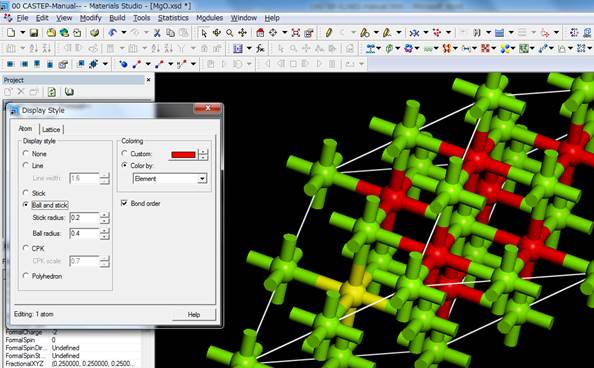
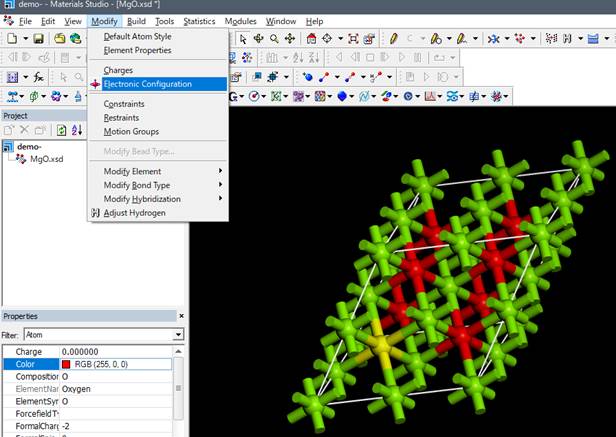
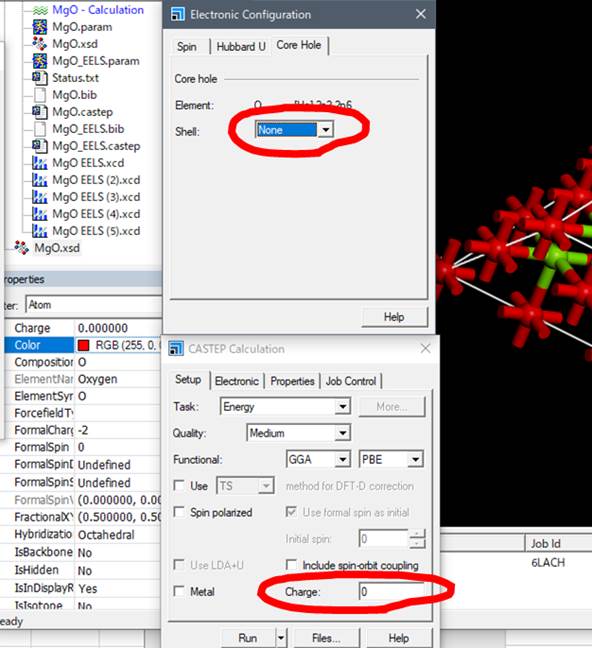
この酸素に内殻空孔を導入するために,選択したままメニューのModify-> Electronic Configurationを選択します.
出てきたWindowの一番右のタブ「Core hole」を選びます.
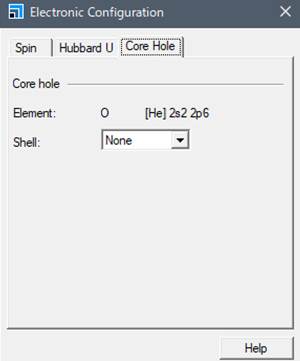
通常は基底状態なのでShellの欄がNoneとなってます.酸素のK端を計算するために1sにcore-holeを導入します.
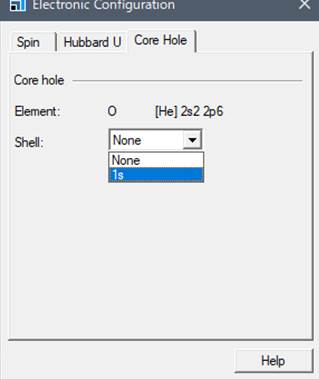
1sを選択したらwindowを閉じます.モデルをみても,見た目では内殻空孔が導入されているのか分かりません.Core-holeを本当に導入できたかを確認するには,Edit->Atom
Selectionを選択し,
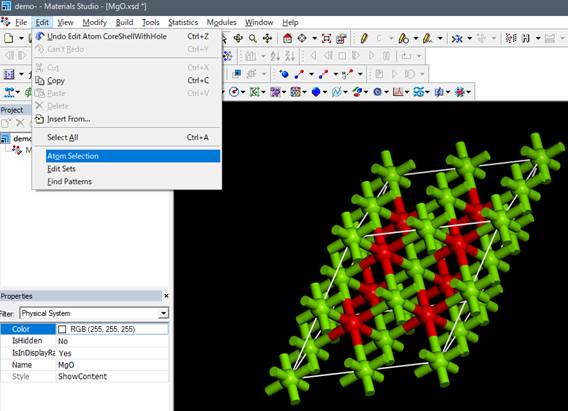
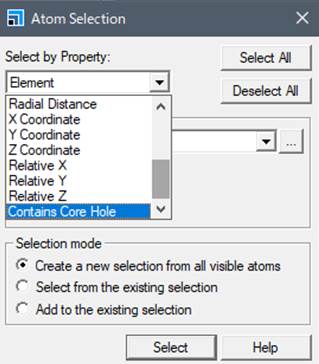
出てきたWindowのSelect by Property:でContain Core Holeを選択して,SelectするとCore Holeを導入した原子が選択されます.わかりやすくするために,内殻空孔を導入した原子をDisplay styleを使って,Ballのサイズを変えておきます.
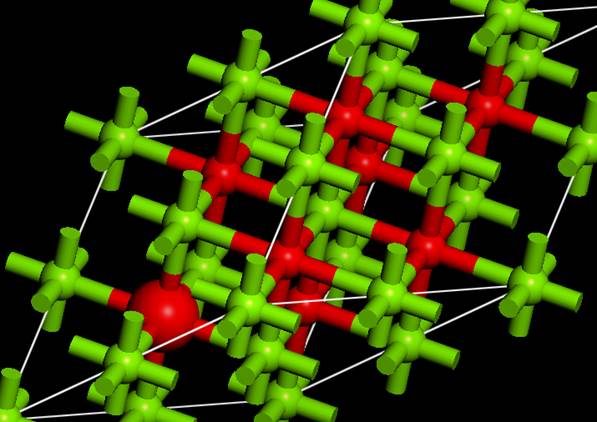
次に,Material Studioはsupercellをつくると対称性がP1に落ちます.P1のままで計算するのは非効率なので,このセルに対称性を与えます.
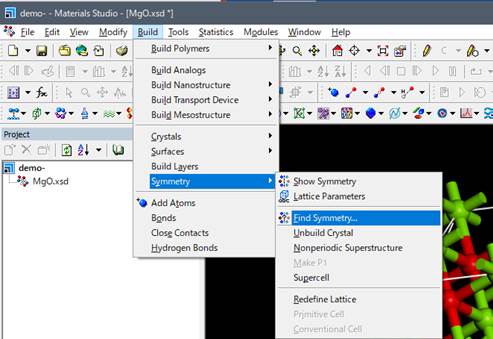
デフォルトの設定でsymmetryを探すと,元素の種類だけで対称性が検索されてしまいます.今回は内殻空孔を導入しており,内殻空孔を導入した励起状態の酸素と,内殻空孔を含まない基底状態の酸素とは区別すべきです.そのためにFind symmetryのwindowの右のタブのOptionsを選択し,CoreShellWithHoleを選択しておきます.
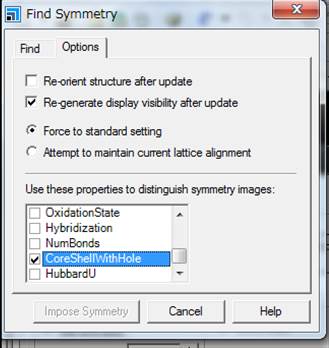
次に,Findのタブにもどり,Find symmetryをします.
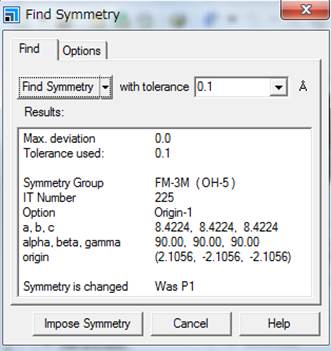
そうすると今回Fm-3mが見つかります.次にImpose symmetryします.

その結果,上図のようなUnit cellを拡張したスーパーセルが作成されます.今回はPrimitive
cellの2×2×2倍したスーパーセルの計算を実施したいので,再びBuild->Symmetry->Primitive
Cellを選択してPrimitive cellにします.
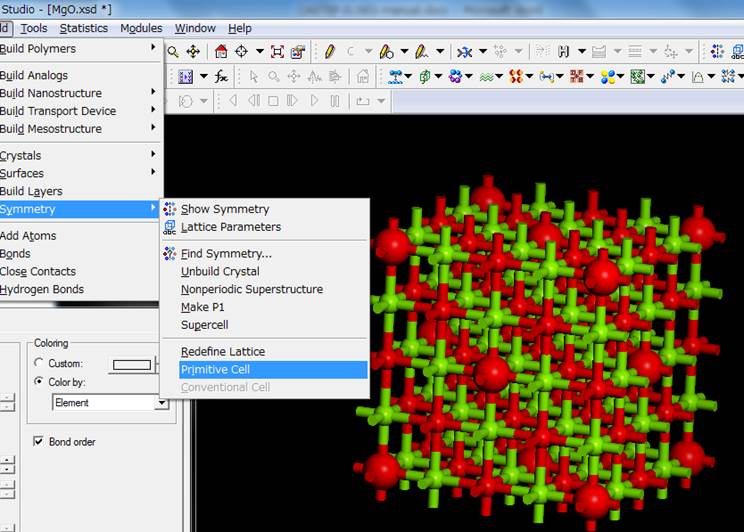
今回は以下のようなセルになりました.つまり,内殻空孔を導入した原子(大きな赤丸)がずれて,スーパーセルの端(もしくは中央)になっていることが分かります.これは,対称性を上げるために,内殻空孔を導入した原子を対称性の高いサイトに移動させたためです.
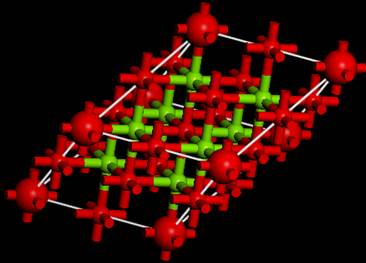
これで,計算を実行するためのセルが完成しました.
3.CASTEPを実行する
次にツールバーからCASTEPのアイコンを選択し,Calculationを選択します.
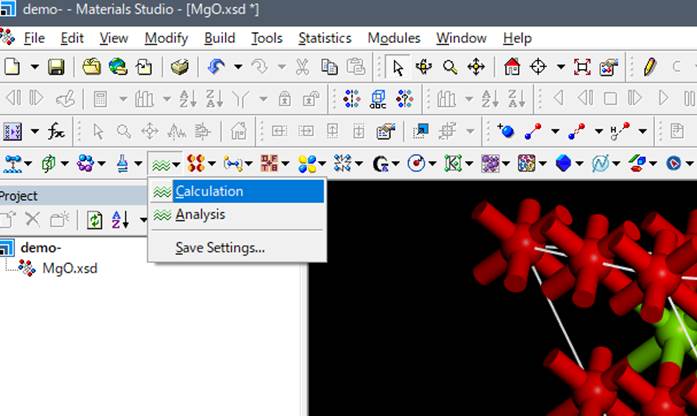
CASTEP CalculationのWindowが立ち上がります.各タブを説明します.まずSet upタブについて,
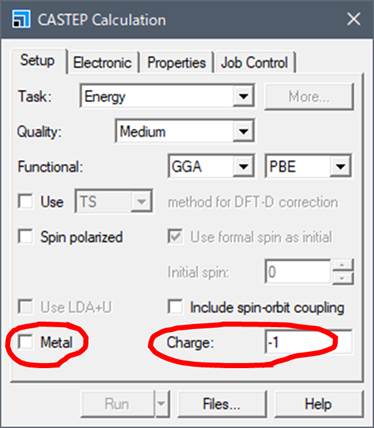
まず,MgOはMetalではないのでMetalのチェックを外します.次に重要なことですが,Chargeを-1にします.これは,Core holeがスーパーセルにある場合にMaterial Studioがセルのチャージを+1にするという勝手な操作を正すためです.Material Studioはマイナス電荷の電子が内殻から抜けて,Core-holeが形成したことでセルとしてプラスになったと勝手に判断します.しかし,ELNES/XANESが反映しているのは内殻電子が伝導帯に入っている電子状態です.つまり,電子の位置が内殻軌道から伝導帯に移動しただけで,スーパーセルのチャージはニュートラルです.これはMaterial Studio版CASTEPのみの問題で,著者がこれまで行ってきたすべての計算(Wien2k,OLCAO,ソース版CASTEP,DV-Xα等)ではすべてニュートラルなセルで計算しています.
そのようなMaterial Studioの操作を正すためにChargeを-1にしておきます.そうすることで入力した-1と,+1が打ち消し合い,結果的にニュートラルな計算を行ってくれます.
次に,Electronicのタブに行きます.
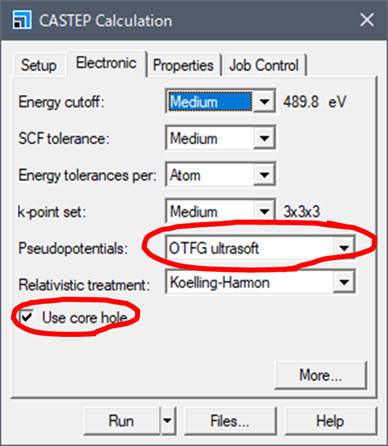
まず,左下の Use core holeを選択します.つぎに,core-holeを導入するためにはPseudopotentialsの種類が On the flyである必要があります.On the flyでは「その場で」pseudopotentialを作成するということです.On the flyで内殻空孔を導入した擬ポテンシャル(励起擬ポテンシャル)を作製し,内殻空孔を導入すべき原子には励起擬ポテンシャルを適用し,他の原子には基底状態の擬ポテンシャルを適用します.
このパネルにEnergy cutoffやk-pointsの設定もできます.ELNES/XANES計算の場合,Energy cutoffやk-pointsは特に理論遷移エネルギーにきいてきます.いくつかの条件(たとえばMedium,
Fine,Ultra-fine)で計算して比較し,計算の精度を確かめる必要があります.
次にPropertiesタブに行きます.
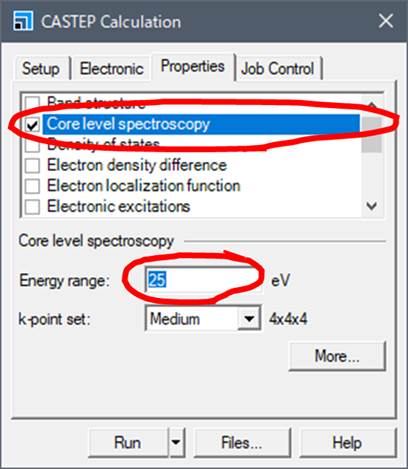
ELNES/XANESを計算するためにCore level spectroscopyを選択します.下にEnergy rangeとk-point setが出てきます.Energy rangeは吸収端から何eV後ろまで計算するかということです.25eVの場合は吸収端から25eVぐらい後ろまでスペクトルを得ることができます.この値を大きくすると非常に計算時間がかかります.
k-point
setはスペクトル計算におけるk点です.Electronicのタブにもk-point setがありましたが,Electronicタブの方は電子構造を計算する(SCF計算)するときのk点で,Propertiesタブの方は,SCF計算の結果をもとにスペクトルを計算するためのk点になり,k点が多いほど滑らかなスペクトルになりますが計算時間がかかります.今回はMediumで行います.
次にJob Controlタブに行きます.

Run in parallelは計算するPCのコア数になります.また,Runtime optimizationがDefaultになってますが,これをSpeedにするとテンポラリーファイルをすべてメモリに格納してくれます.メモリを多く積んだPCであれば計算がかなり高速になります.
* 以前のMaterials
Studioではサーバーに計算結果を残す設定をしていました.しかし,最近のMaterials Studioではその操作は不要です.
4.ELNES/XANES計算結果をみる
しばらくすると計算が終了し,Materials studioが以下のような表示になります.
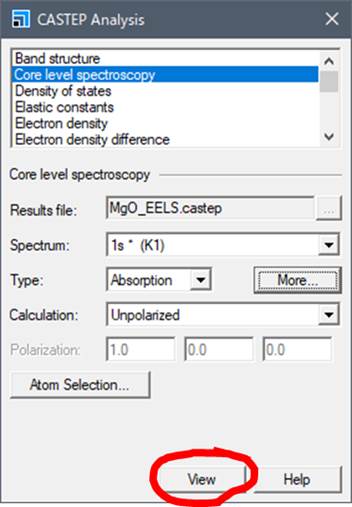
次にCastepボタンのAnalysisを選びます(下図).
次に,計算に使ったMgO.xsdを選択して,内殻空孔を導入した酸素(大きな赤い酸素)を選択し,CASTEP AnalysisのWindowでCore level spectroscopyを選択します.
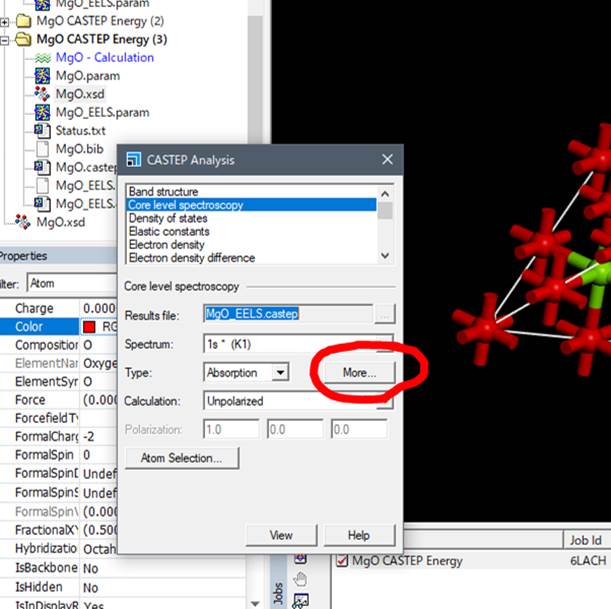
出てきたCASTEP AnalysisのCore level spectroscopyのところで,上記のようにMoreを選択します.2014年版からはこの辺が変更されてます.Moreを選択すると以下のようなwindowが開きます.
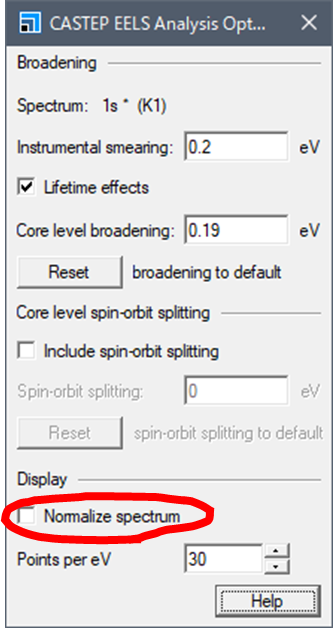
ここでブロードニングの幅などを設定できます.下のほうに"Normalize spectrum"というチェックポイントがあります.ここを外します.
以前のMaterials Studioでは計算スペクトルのピークトップをすべて1.0にノーマライズしてました.最近のMaterials Studioではここのチェックを外すことでノーマライズしない,生の計算結果が得られます.
*ただし,以前のMaterials
Studioではテキスト形式で出力されていた,case_EELS.elnesファイルはバイナリ化されてcase_EELS.elnes_binとなっており,ユーザーは中身を見ることができません.
次に先ほどのCASTEP AnalysisのWindowにもどります.他に,TypeでAbsorptionとEmissionが選択でき,Calculationで,UnpolarizedとPolarizedを選択することができます.ELNES/XANESはAbsorptionです.また注目する原子の電子構造に異方性がある場合,スペクトルに方位依存性があらわれます.異方性がある場合は,Polarizedにして,100や010,001にしてViewすると各方位に対するスペクトルを得ることができます.MgOの場合,酸素もMgにも異方性は無いのでUnpolarizedにして,viewを押します.
Viewを押すとMaterials StudioのWindow内で選択した酸素原子のO-K端ELNES/XANESを表示します.今回は以下のようなスペクトルが得られました.
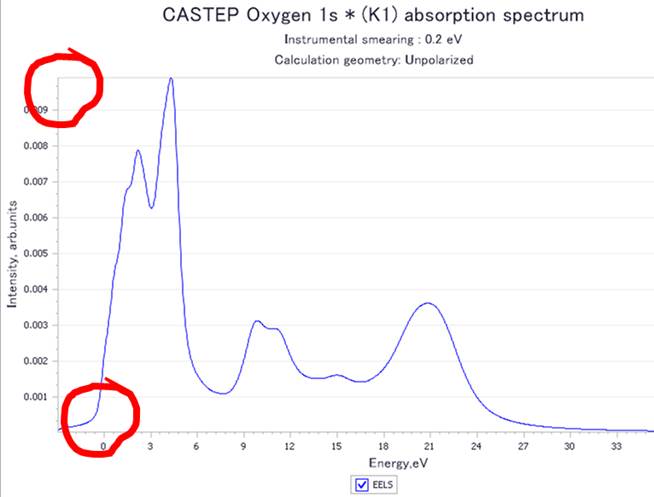
横軸に注目すると,ゼロからピークが立ち上がっていることが分かります.これは遷移した電子が伝導帯下端にいることを示してます(計算がきちんとできている).次に縦軸上部の値をみてみます.ノーマライズされていない値(非1)になっていることが分かります.
2014年版ではノーマライズを回避するために,計算の元データにアクセスする必要がありましたが,前述のノーマライズのチェックボタンをはずして出力すれば,ノーマライズ無しのデータを得ることができます.
このグラフデータを使用するために,グラフエリアで右クリックからCopy(Ctrl+c)をします.
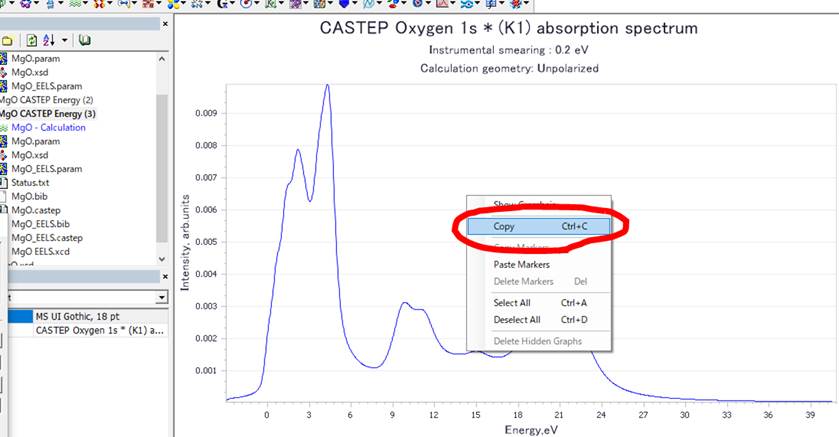
そして,そのままExcelなどのグラフソフトや,テキストエディターを立ち上げてペースト(Ctrl+v)します.
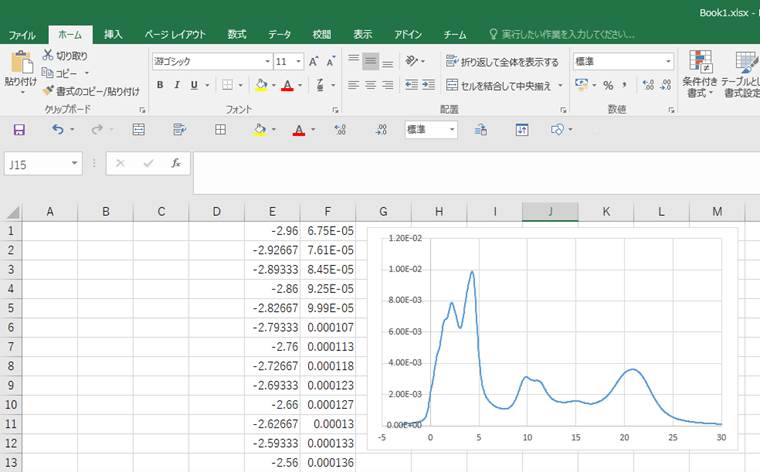
Materials StudioのWindow内で観ていたグラフをつくることができます.また,縦軸がノーマライズされてないことも分かります.
5.遷移エネルギーの計算方法
最後にCASTEPを用いた遷移エネルギーの算出方法を説明します.詳細は論文T.
Mizoguchi et al. J. Phys.: Cond. Matter.,21 (2009) 104204-1-6.をご参考ください.
まず,基底状態を計算します.先ほどのMgO 16原子のモデルで,その内殻空孔を戻し,Chargeもゼロにします.
遷移エネルギーの計算のためには基底状態のエネルギーだけあればよいので,基底状態のスペクトルを出す必要もありません.基底状態はSCF計算のみ実行します.
ここまでで,CASTEPによる遷移状態(Excited/Final state)と基底状態(Ground state)の全エネルギーが計算されています.内殻軌道もすべて取り扱う全電子計算法で全エネルギーを計算した場合はこの二つの全エネルギーのみで,遷移エネルギーを計算することができます.一方で,CASTEPは内殻軌道を計算しない擬ポテンシャル法なので,内殻軌道の寄与を加味する必要があります.具体的には,CASTEPを用いて遷移エネルギーの計算には以下の6つの値が必要になります.
1.基底状態の全エネルギー Total energy @ Ground state(case.castepファイル中ごろ)
2.遷移状態の全エネルギーTotal energy @ Final state(case.castepファイル中ごろ)
3.基底状態の単原子の全エネルギー Single atom total energy @ Ground state(case.castepファイル初めあたり)
4.遷移状態の単原子の全エネルギー Single atom total energy @ Final state(case.castepファイル初めあたり)
5.基底状態の単原子の価電子エネルギー Single atom valence energy @ Ground state(case.castepファイル初めあたり)
6.遷移状態の単原子の価電子エネルギーSingle atom valence energy @ Final state(case.castepファイル初めあたり)
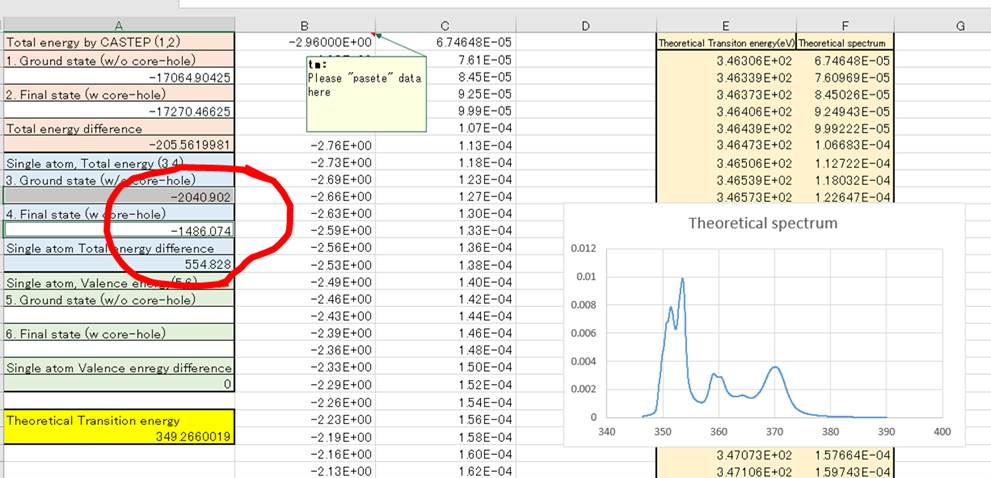
計算するのに役立つエクセルファイルを公開しております(こちら).エクセルでそのまま開くと以下のようなものになってます.
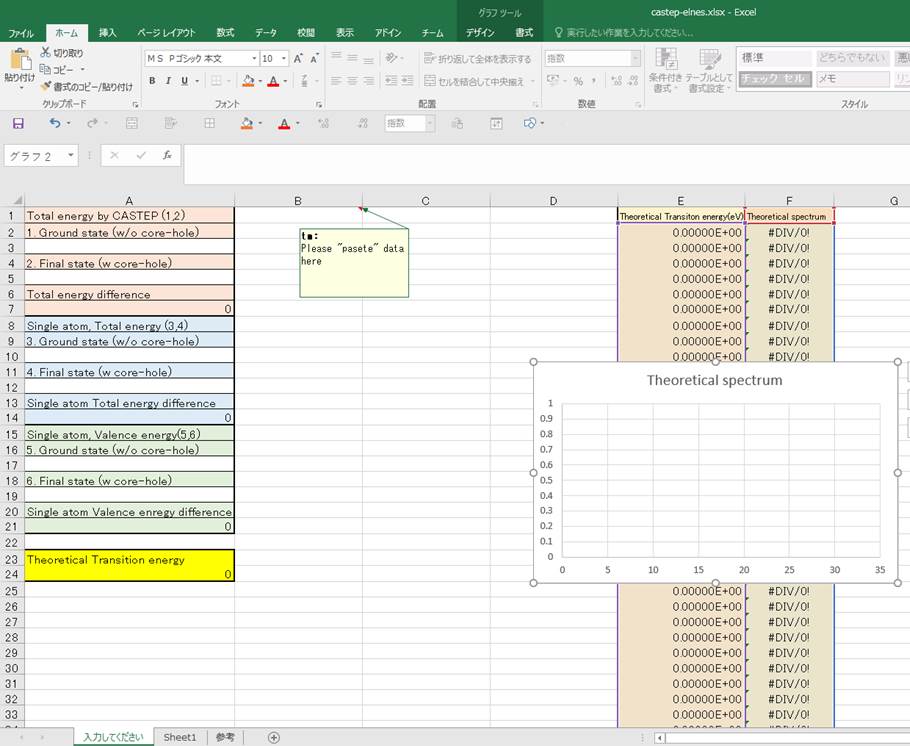
英語になってますが,E列のTheoretical transition energyが遷移エネルギー(x軸),Theoretical spectrumがスペクトル(y軸)になります.計算としては,上記参考文献にある
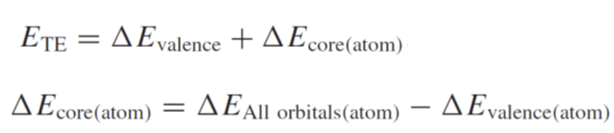
を行っています.ETEが遷移エネルギーで,上記のエネルギー1〜6を使うと
ΔEvalence=
2 - 1
ΔEcore(atom)=
(4 – 3) – (6 – 5)
*ΔEAll
orbitals (atom)= 4 – 3,ΔEvalence
(atom)= 6 - 5
ということになります.これは,CASTEPで計算されていない内殻の寄与を,孤立単原子の内殻で加味するということになります.内殻電子はエネルギー的にも空間的にも局在しているため,この近似は多くの場合うまく働きます.
まず,Materials Studioのグラフデータをコピーして,B1のカラムを選択した状態で,ペーストします.そうすると
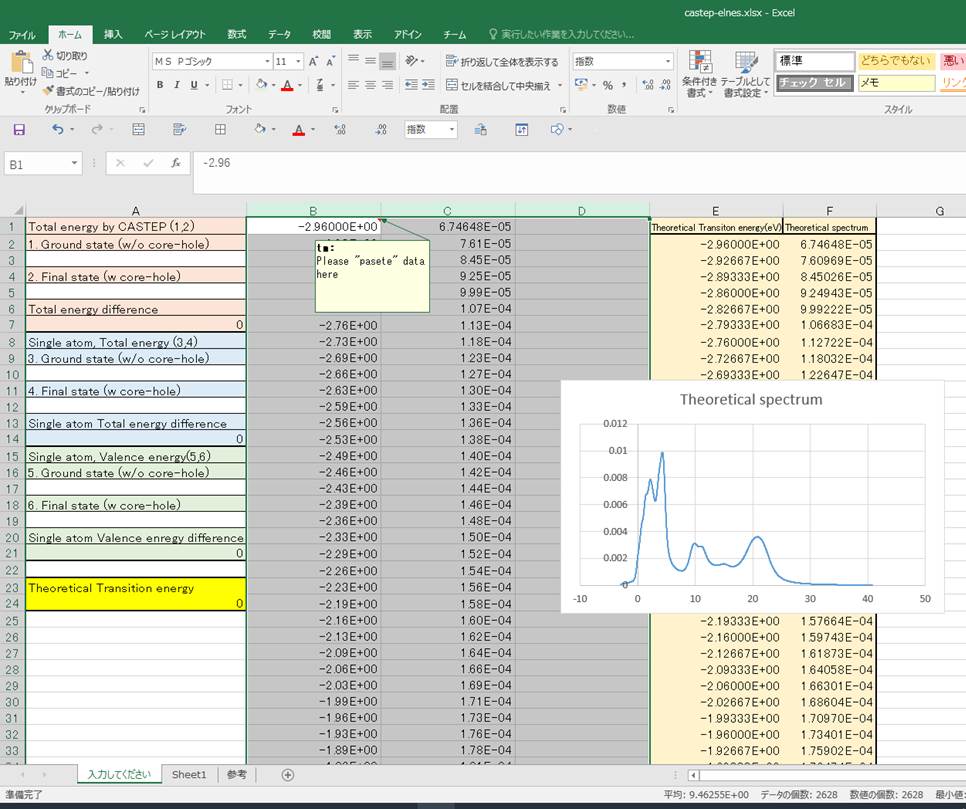
このようになるかと思います.次に,エネルギーを入力していきます.
1.基底状態の全エネルギー Total energy @ Ground state(case.castepファイル中ごろ)
2.遷移状態の全エネルギーTotal energy @ Final state(case.castepファイル中ごろ)
基底状態を計算したMgO-gs.castepファイル(.castepの前は各計算時の名前)の中ごろに以下のようにSCFでの全エネルギー―の値が出てます.
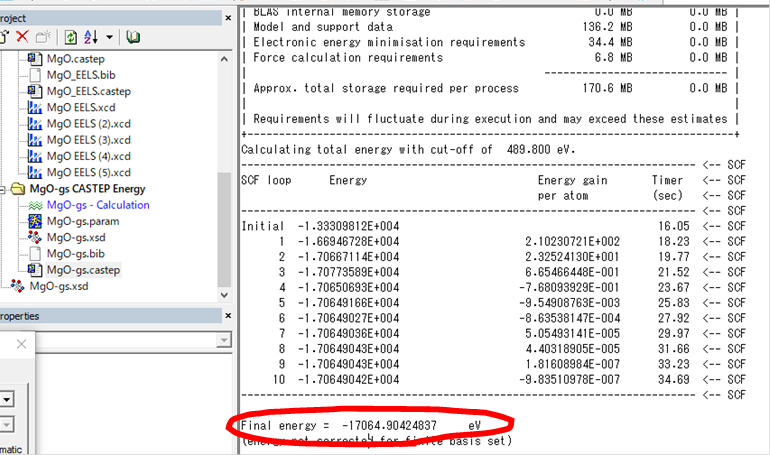
このFinal energyが基底状態の全エネルギーとなります.同様に励起状態の.castepファイルから励起状態の全エネルギーが分かります.二つの値をエクセルの1.,2.のセルに入力します.
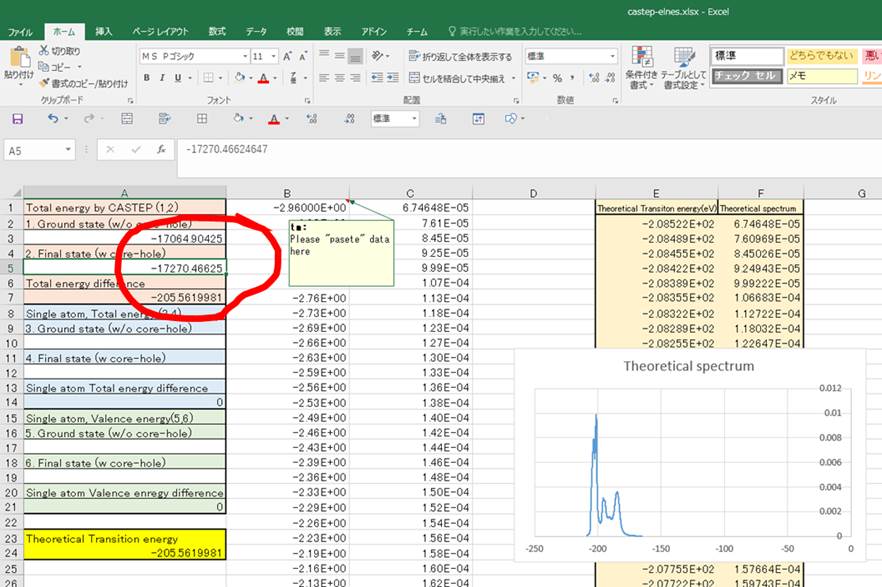
この段階ではTheoretical transition energy(黄色セル)が-205.56eVぐらいになってしまってますが,これは物理的に正しいので先に進みます.
次に,基底状態および基底状態,遷移状態のcase.castepファイルの上の方に,単原子の全電子エネルギーが出力されてます.
3.基底状態の単原子の全エネルギー Single atom total energy @ Ground state(case.castepファイル初めあたり)
4.遷移状態の単原子の全エネルギー Single atom total energy @ Final state(case.castepファイル初めあたり)
基底状態のcase.castepでは以下のように出力されてます.
遷移状態のcase.castepは以下のような出力されてます.
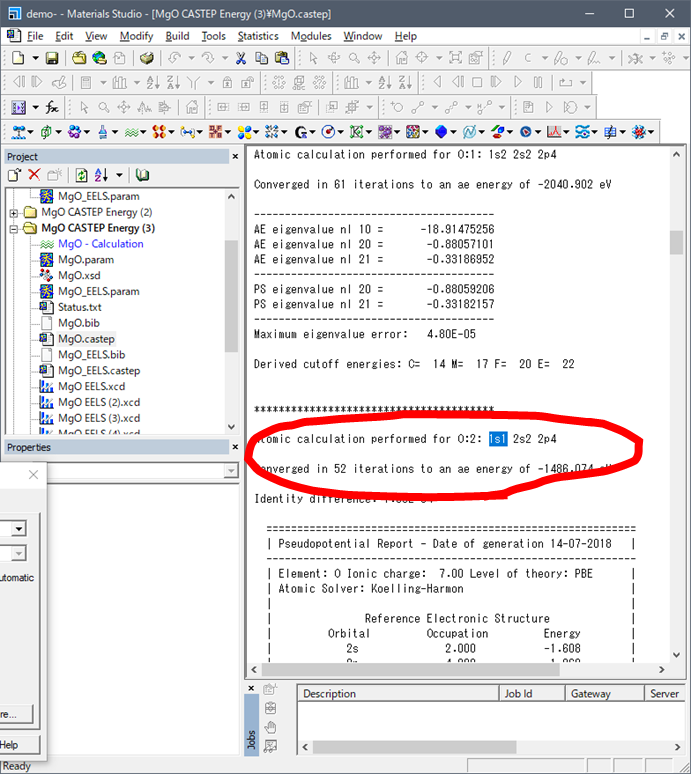
遷移状態では酸素の1s電子数が1s1になっていることが分かります.これらの値をエクセルの3., 4.に入力します.
遷移エネルギーが350eVぐらいになって,だいぶ実験値に近づいてきました.
次に,単原子の価電子エネルギーです.同様にcase.castepの初めに出力されてます.
5.基底状態の単原子の価電子エネルギー Single atom valence energy @ Ground state(case.castepファイル初めあたり)
6.遷移状態の単原子の価電子エネルギーSingle atom valence energy @ Final state(case.castepファイル初めあたり)
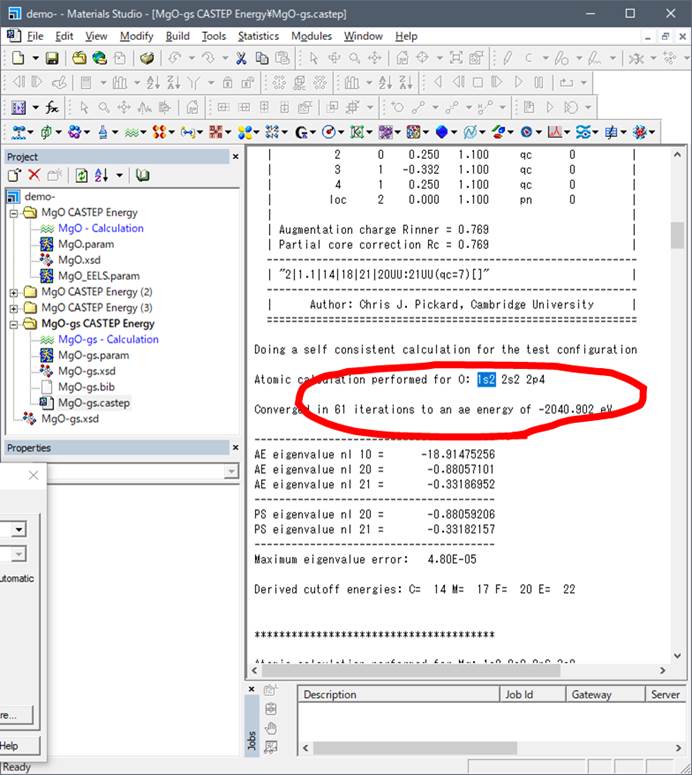
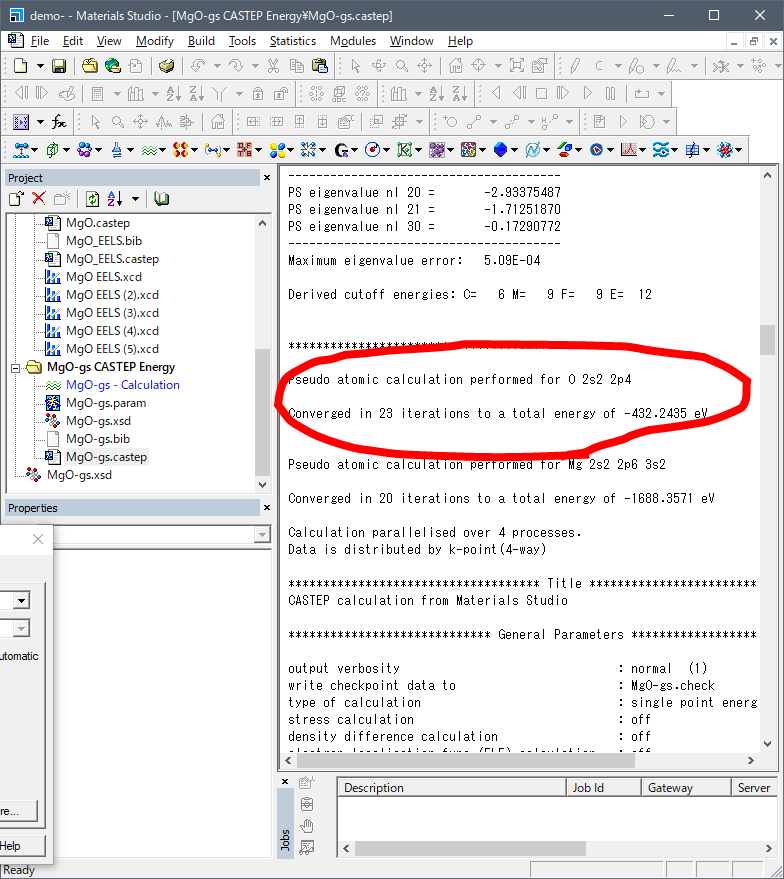
Case.castepの"Pseudo atomic calculation performed for"を探します.そうすると基底状態では以下のような場所が見つかります.
O 2s2 2p4ということで,Oの内殻(1s2)を除いたValence
energyがここに出力されています.
遷移状態では以下のようになっています.
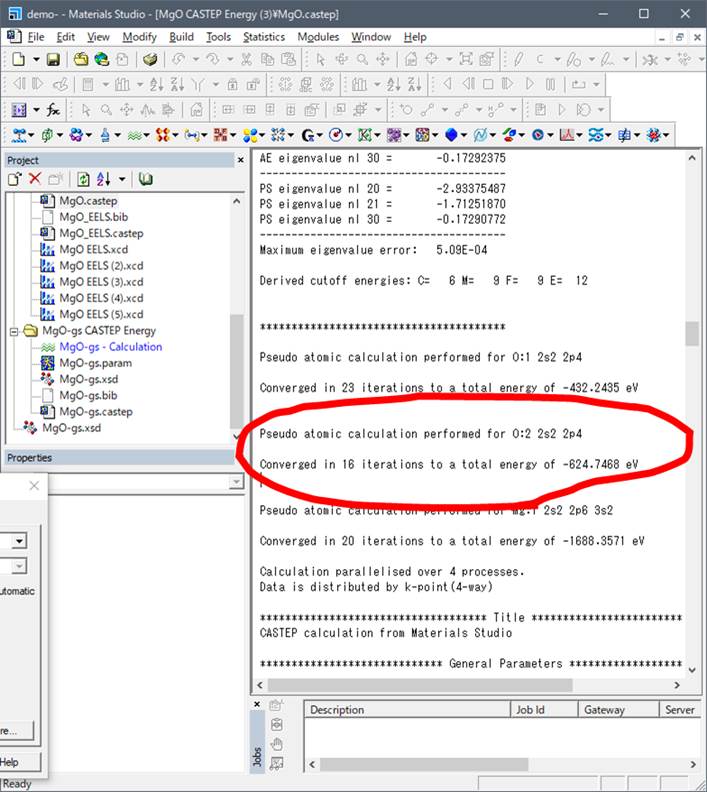
内殻空孔を導入したのはO:2でしたので,O:2のValence
energyを使います.これら基底状態,遷移状態のValence energyをエクセルの5.,6.に入力していきます.
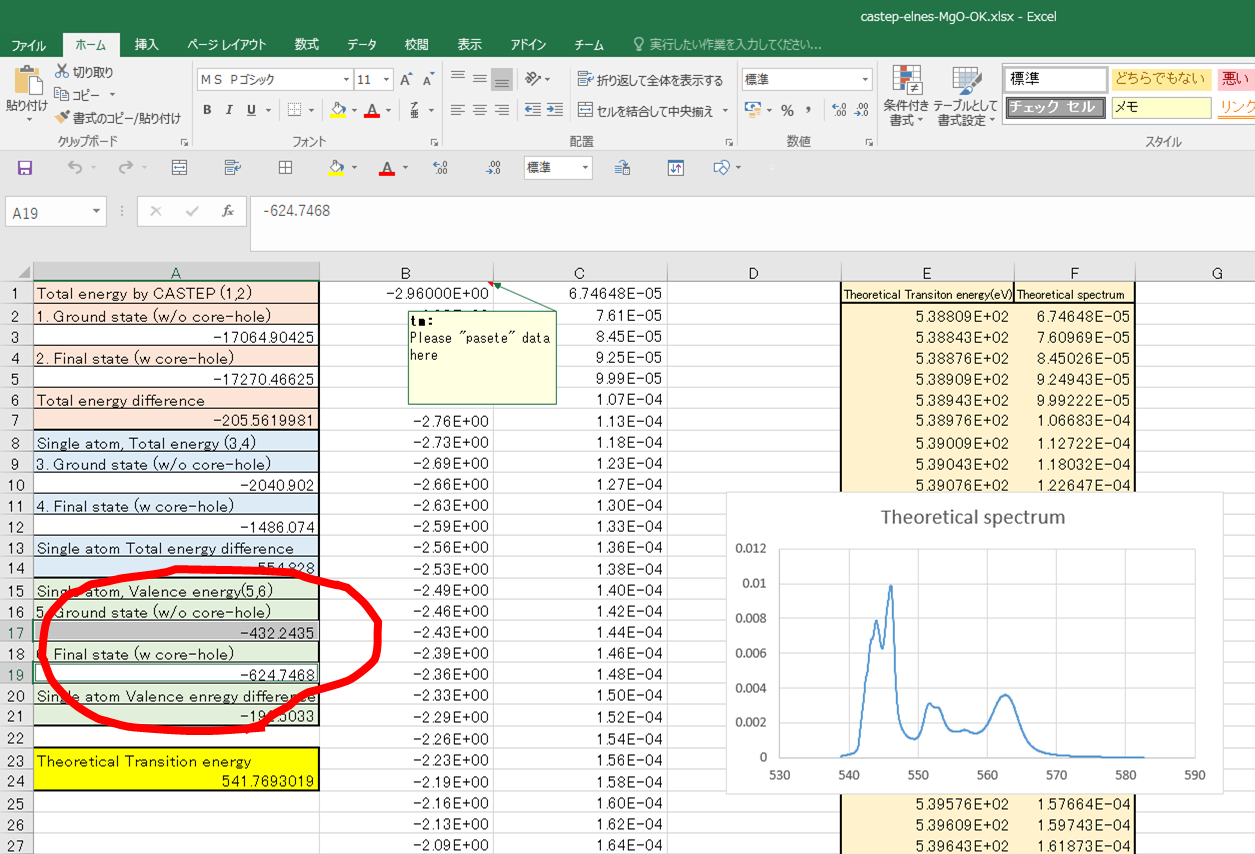
その結果,遷移エネルギーが541.8eVと求められました.この値が計算で得られたELNES/XANESスペクトルの立ち上がりのエネルギーになります.
ここで得られたスペクトルが,MgO 16原子のスーパーセルを用いて計算されたO-K端の計算スペクトル.ということになります.
以上のようなステップにより,CASTEPを用いてELNES/XANESの理論計算を行うことができます.
参考文献
CASTEPについて
"First
principles methods using CASTEP"
S.
J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson
and M. C. Payne,
Z.
Kristallogr., 220, 567–570 (2005).
CASTEPを用いたELNES/XANES理論計算について
"First
principles calculation of spectral feature, chemical shift, and absolute
threshold of ELNES and XANES using plane wave pseudopotential method"
T.
Mizoguchi, I. Tanaka, S Gao, and C.J. Pickard,
J.
Phys.: Cond. Matter.,21 (2009) 104204-1-6.
CASTEPを用いた液体のELNES/XANES理論計算について
"
An estimation of molecular dynamic behaviour in a liquid using core-loss
spectroscopy"
Y.
Matsui, K. Seki, H. Hibara, T. Mizoguchi
Scientific Reports, 3 (2013)
3503-1-7. [free access]
CASTEPを用いた気体のELNES/XANES理論計算について
"Estimation of the molecular vibration
of gases using electron microscopy"
H. Katsukura, T. Miyata, M. Shirai, H.
Matsumoto, and T. Mizoguchi
Scientific Reports, 7 (2017), 16434-1-9. [free access]
ELNES/XANES第一原理計算について
"Basics
and Applications of ELNES calculation" (Invited Review)
H.
Ikeno and T. Mizoguchi
Microscopy,
66 (2017) 305–327. [free
access]
"Theoretical
ELNES: one particle and many particle calculations"(Invited Review)
T.
Mizoguchi, W. Olovsson, H. Ikeno, and I. Tanaka
謝辞:当研究室の大学院生,大阪府立大学・池野豪一先生,京都工繊大・小林久芳先生,日本電子・西藤哲史氏,ダッソーシステム・A. Chatterjee博士,Cambridge Univeristy・Chris J. Pickard先生,Fudan University・S. P. Gao先生,京都大学・田中功先生.