First principles calculation of ELNES/XANES using CASTEP
Teruyasu Mizoguchi
English Ver.2021812
Institute of Industrial Science,
The University of Tokyo
[Mizoguchi Research
Laboratory]
This page describes the
first-principles calculation method of ELNES/XANES using the CASTEP code,
which is the first principles pseudopotential method. We will explain a
technique using a GUI called Materials
Studio of the Dassault system. In this paper, the O-K end of
magnesium oxide (MgO) is calculated as an example. CASTEP uses files with .cell, .param, .castep
extensions, but the name before the extension is case.castep
in this article.
If
you are License holder of CASTEP source code, you may already have .elnes
file. Then, please go to "Calculate Theoretical transition energy" below.
1.Constructing
supercell using Materials Studio
The first step is to create a
model of MgO, which can be obtained from Materials Project, Mat navi, or other databases, or from a space group and atomic
coordinates. In this case, go to the Materials Studio menu File-> Import, go
to the Structures shortcut, and call MgO.msi in Metal-oxides.
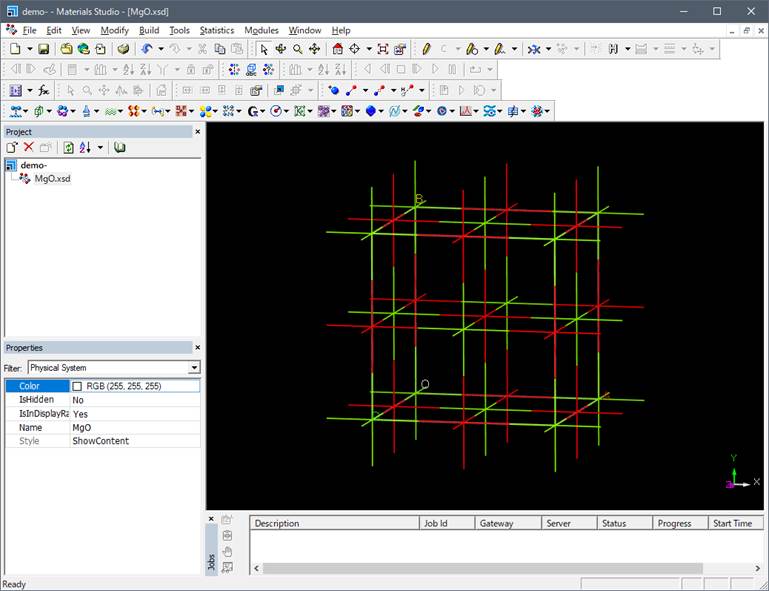
In the above figure, oxygen is
shown in red and Mg in green.
This structure is a unit cell of the rock-salt type structure, so it is changed
to a primitive cell to reduce the computational cost.
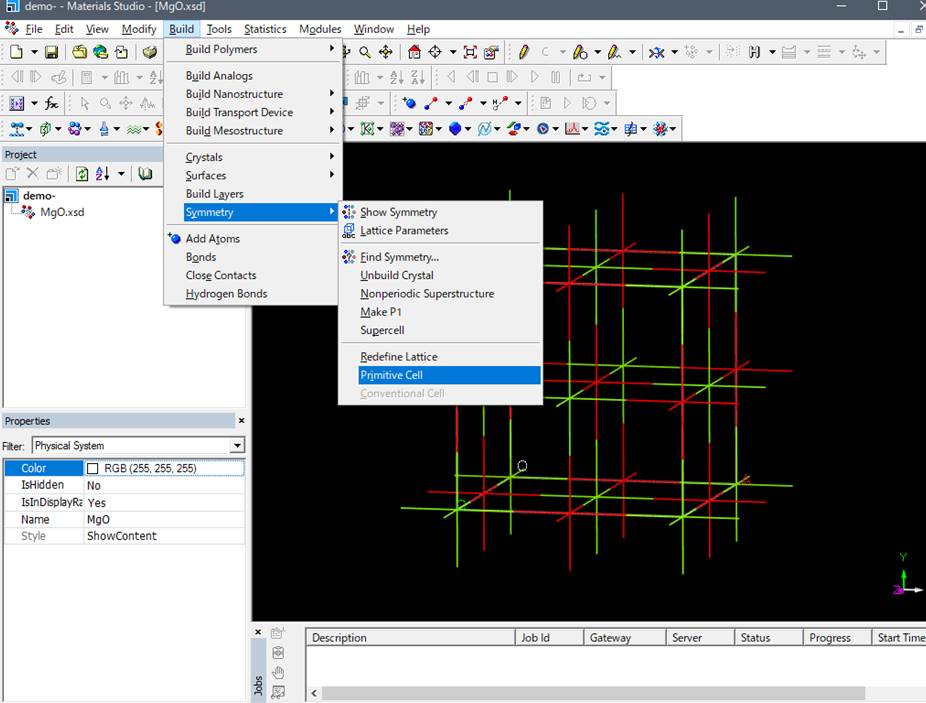
A primitive cell of MgO consisting of two atoms, Mg 1 O 1, is
created. Ground-state calculations (band structure, density of states, etc.)
are performed using the primitive cell with an increased number of k-points.
However, a core-hole effect, which is essential for the ELNES/XANES
calculations, cannot be calculated accurately using this primitive cell.
The core-hole affects to the electronic state very much. In the case
of band structure calculation, the cell to be calculated is extended (periodic
boundary condition), so that interactions occur between the
core-hole-introduced atoms (excited atoms) in the neighboring cells (see the
figure below).
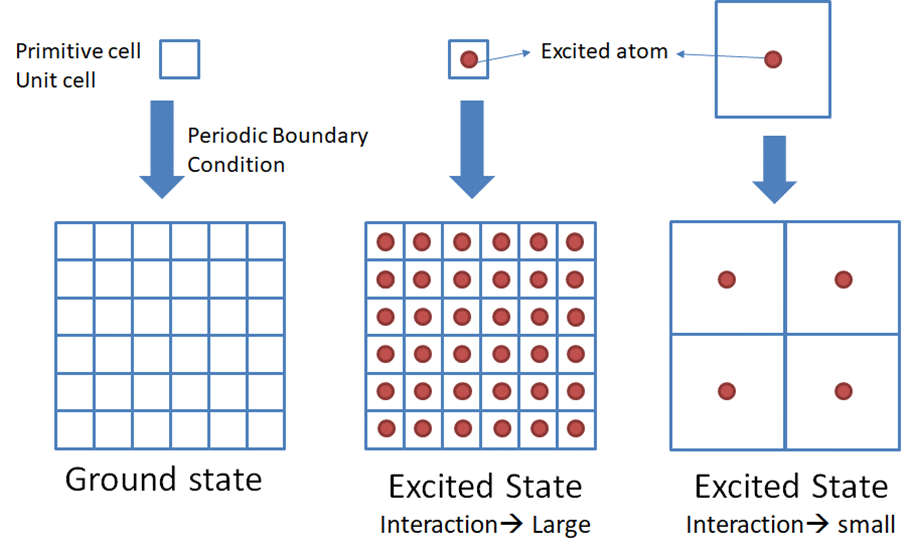
Thus, we need to use an
extended cell (supercell) instead of a small primitive (or unit) cell to reduce
the interactions between atoms with core-hole. Previous studies have shown that
it is necessary to keep 1 nm (10 Å) separation between atoms with core-hole.
Here, for rapid computation, we use a 2x2x2 super cell, which is a 2x2x2 expansion
of the MgO primitive cell as shown below. (Officially, you need to use more
than 54atoms supercell for the accurate simulation of O-K edge of MgO).
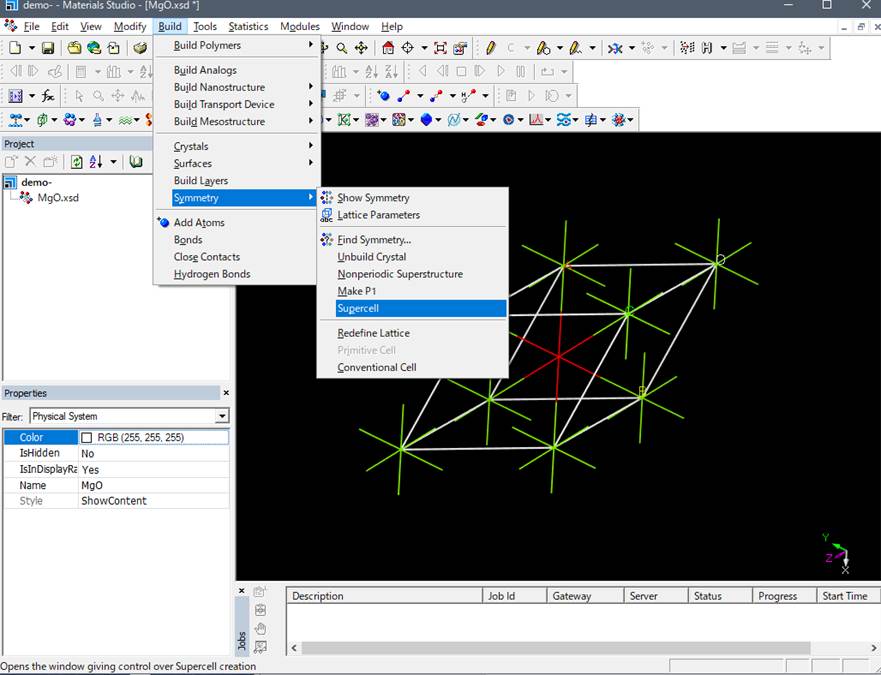
For instance,
a 16-atom supercell can be generated by multiplying by 2 x 2 x 2, as shown
below.
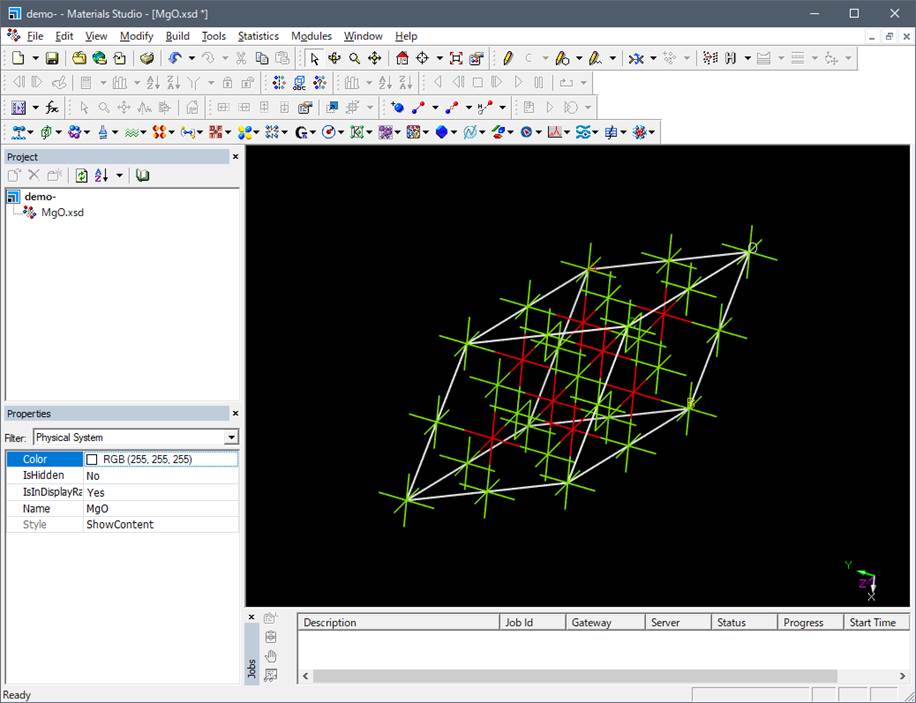
Here, it should be mentioned
that the symmetry of the crystal goes to P1
(no symmetry) when you construct the supercell using the Materials Studio.
2.Introduction
of core-hole
Since we are going to calculate
the K-edge of oxygen, we will choose an oxygen atom by click. It doesn't matter
which oxygen you choose. In the "Line" view, it is difficult to
understand, so right-click and select "Ball and stick" in the Display
style, and select one oxygen atom. In this example,
the oxygen atom in the lower left corner of the cell (yellow) is selected.
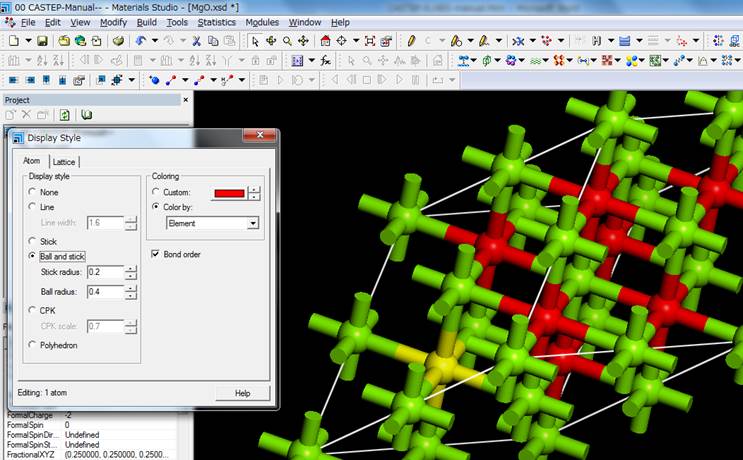
To introduce an inner-shell
vacancy into this oxygen, select Modify-> Electronic Configuration from the
menu while it is selected.
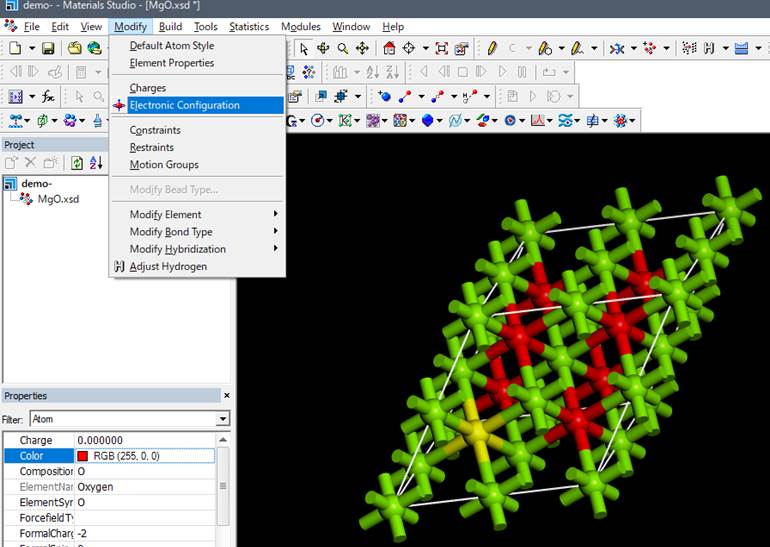
Select the rightmost tab
"Core hole" in the window that appears.
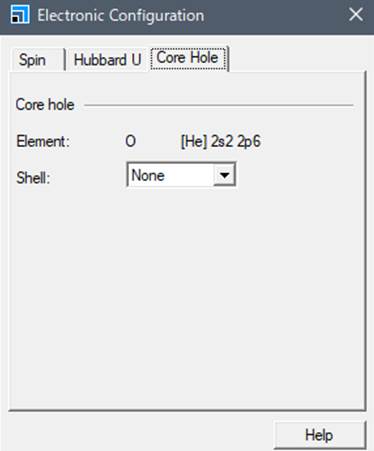
Normally, the "Shell"
is in the ground state, so the "Shell" column is set to None. To
calculate the K-edge of oxygen, we introduce core-hole in 1s.
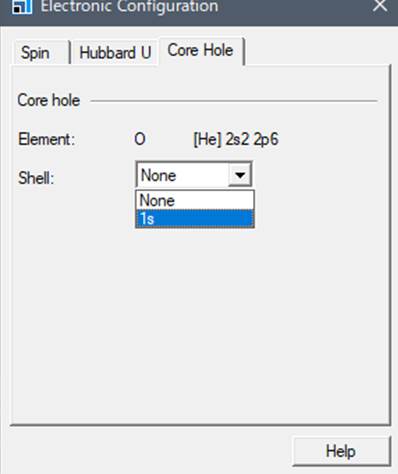
After selecting 1s, close the
window. To check whether the core-hole has really been introduced, select
Edit->Atom Selection and click
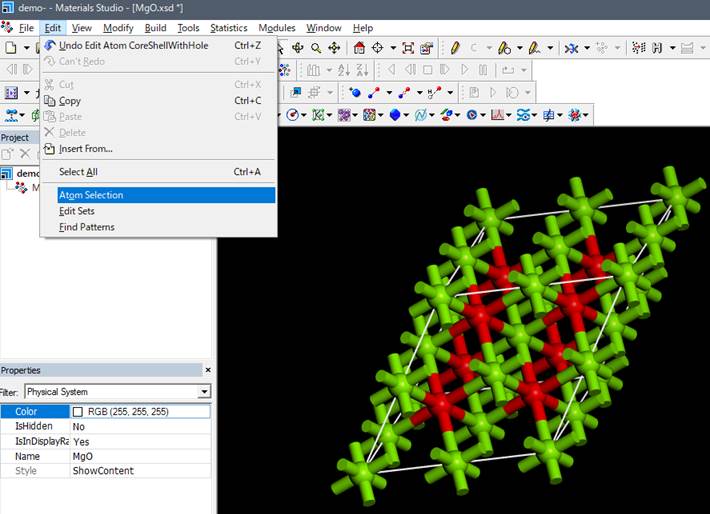
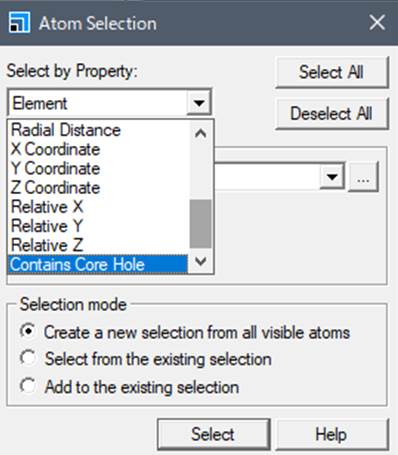
In the window that appears,
select "Contain Core Hole" in the Select by Property: field, and then
select the atom with the core hole. To make it easier to understand, we will
use the Display style to change the size of the ball for the atom with the core
hole.
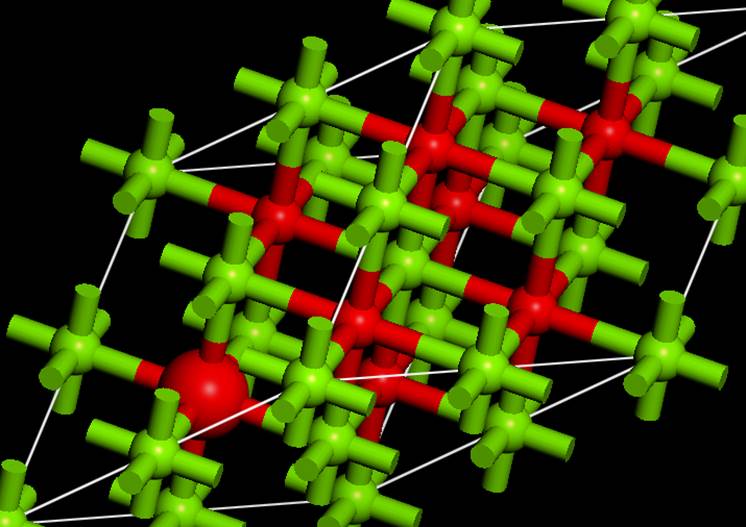
Next, Material Studio set the
symmetry to be P1 when you make the supercell, and since it is inefficient to
compute with P1, we will give symmetry to the supercell.
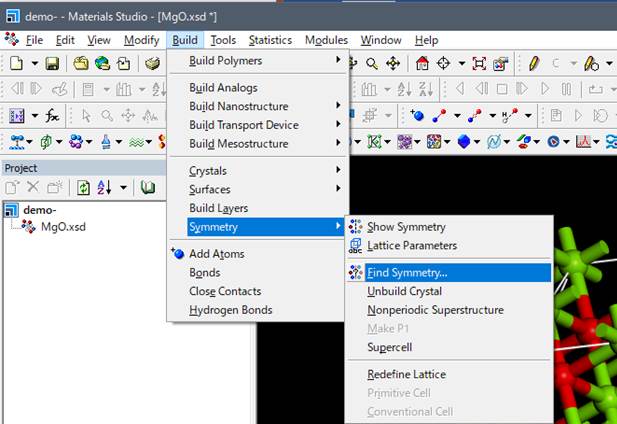
Here, you have
to be careful because the symmetry will be searched only by element type
when you use the default settings of "Find symmetry". In this case,
we are introducing core-hole, and we should distinguish between the excited
state oxygen with core-hole and the ground state oxygen without core-hole. For
this, select Options on the right tab of the Find symmetry window, and select CoreShellWithHole.
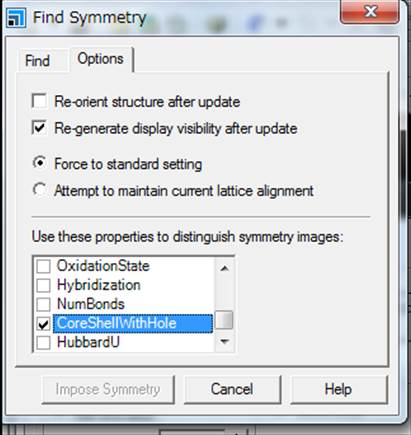
Then, go to "Find
symmetry".
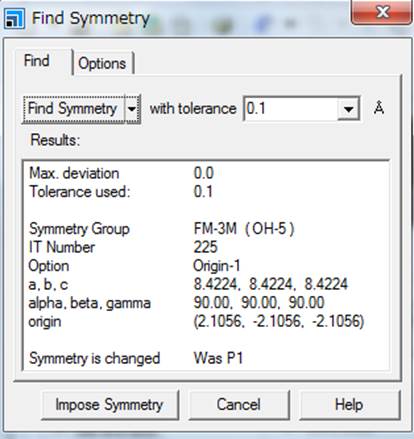
You may get Fm-3m symmetry,
then impose symmetry.

As a result, a super cell that
extends the Unit cell is created as shown in the above figure. This time, we
want to perform the calculation of a super cell that is 2 x 2 x 2 of the
primitive cell, so select Build->Symmetry->Primitive Cell again to make
it the (expanded) primitive cell.
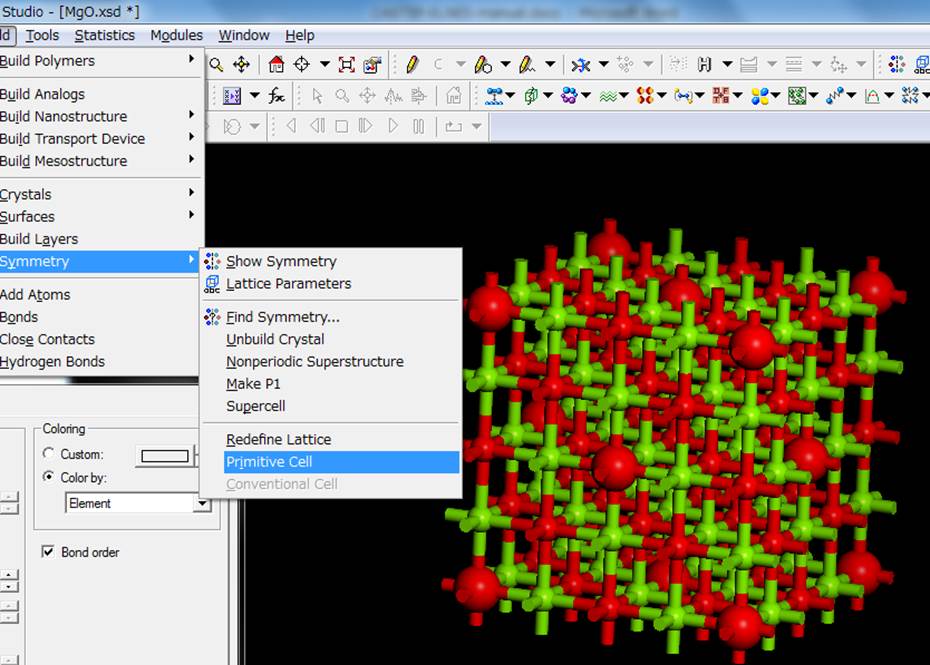
Then you can obtain the
following supercell.
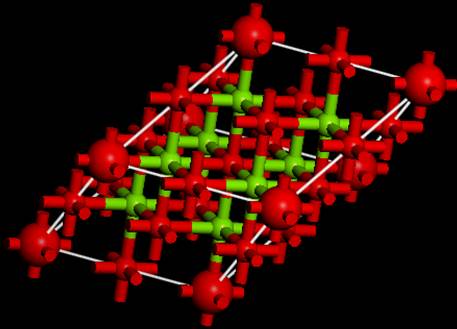
Now, you have the supercell to
be simulated!!
3.Running
CASTEP
Next, select the CASTEP icon
from the toolbar and select Calculation.
The CASTEP Calculation window
will be launched. Each tab is explained below. First, let's look at the Set up
tab.
First, since MgO is not Metal, uncheck Metal.
(Here, it depends on your
Materials Studio version. If your Materials Studio is 2019 or newer, I have confirmed it is OK. But it is old version, like 2018, you
need to set Charge to -1. This is old Materials studio’s bug!! This operation
is necessary to correct Material
Studio's arbitrary setting of the cell's charge to +1 when the core
hole is in the supercell. This bug
was fixed in at least Ver.2019-2021!! )
Next, go to Electronic tab
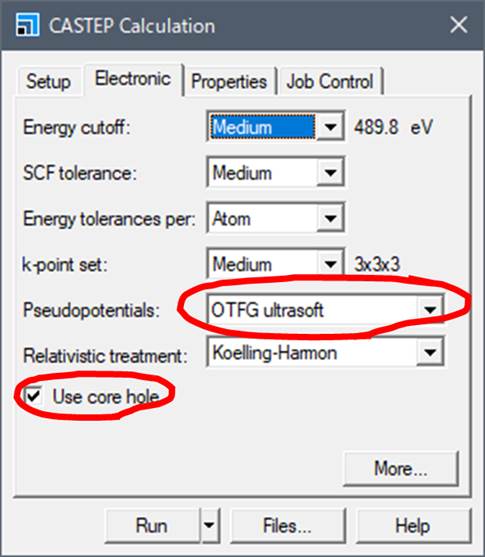
First, select Use core-hole in the lower left corner. In order
to introduce a core-hole, the type of pseudopotential must be On the fly (OTFG), which means that the pseudopotential is
created "on the fly".
In the case of ELNES/XANES calculations, the Energy cutoff and
k-points are particularly important for theoretical transition energies. It is
necessary to compare calculations under several conditions (e.g., Medium, Fine,
Ultra-fine) to verify the accuracy of the calculations.
Lets go to Properties tab.
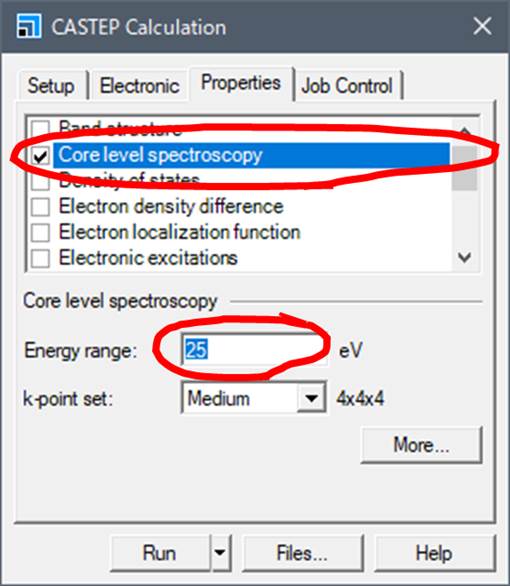
Select Core level spectroscopy to calculate ELNES/XANES. The
Energy range is the number of eV behind the absorption edge. 25eV means you can
get the spectrum up to 25eV behind the absorption edge. If you increase this
value, the calculation time will be very long. Actually,
ELNES simulation is almost 10~100 times time consuming than SCF
simulation!!
k-point set is a k-point for the ELNES/XANES calculation.
k-point set is also available in Electronic tab, but
Electronic tab has k-points for SCF calculation, and Properties tab has
k-points for calculating the spectrum based on the result of SCF calculation.
The more k-points you have, the smoother the spectrum will be, but it will take
more time to calculate.
Next, lets
go to Job control tab.
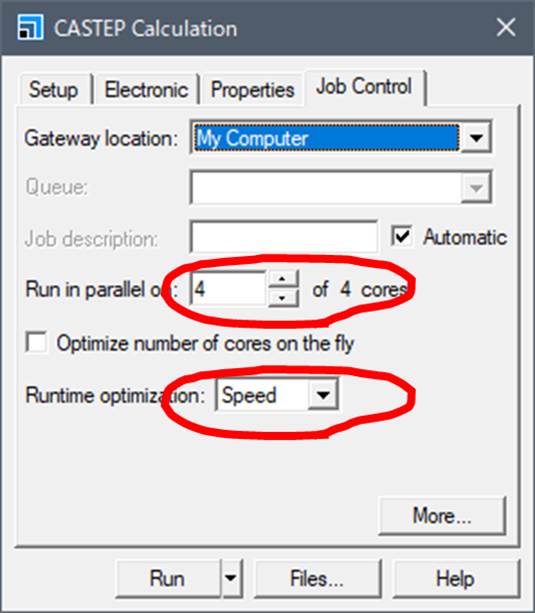
Run in parallel is the number of cores on the PC. Also, Runtime
optimization is set to Default, but if you set it to Speed, all temporary files
are stored in memory.
4.Visualization
of ELNES/XANES spectrum
After the simulation was
finished, Materials studio will be like below
Then, lets go to Analysis of the Castep
button(as below)
Next,
select the MgO.xsd file used for the calculation, select the oxygen that
introduced the core-hole (the big red oxygen), and select Core level
spectroscopy in the CASTEP Analysis window.
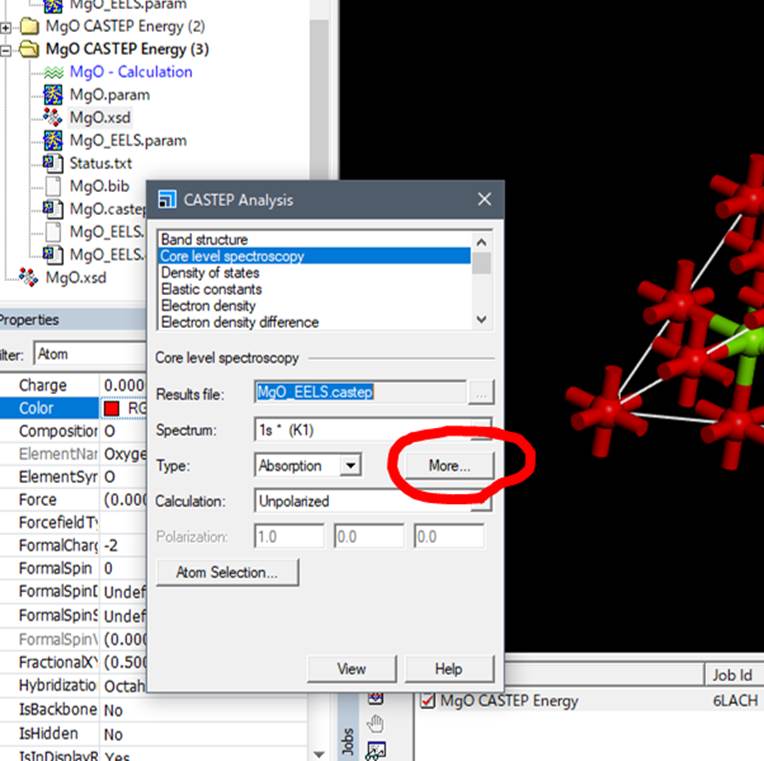
In the
Core level spectroscopy section of CASTEP Analysis, select More as shown above.
Then, the following window is launched.
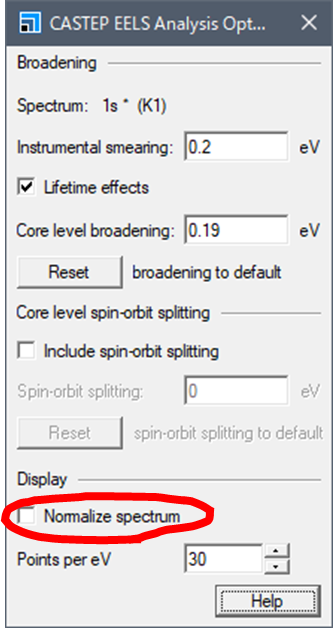
Here you can set the width of the
broadening. At the bottom,
there is a checkpoint called "Normalize spectrum". Remove this checkpoint. Otherwise the spectral intensity was normalized by the
topmost peak of your spectrum.
Next,
go back to the CASTEP Analysis window. In addition, you can select Absorption
or Emission in Type, and Unpolarized or Polarized in Calculation. If there is
an anisotropy in the electronic structure of an atom of interest, the spectrum
will show an orientation dependence. In the case of MgO, there is no anisotropy
in either oxygen or Mg, so select Unpolarized and press view.
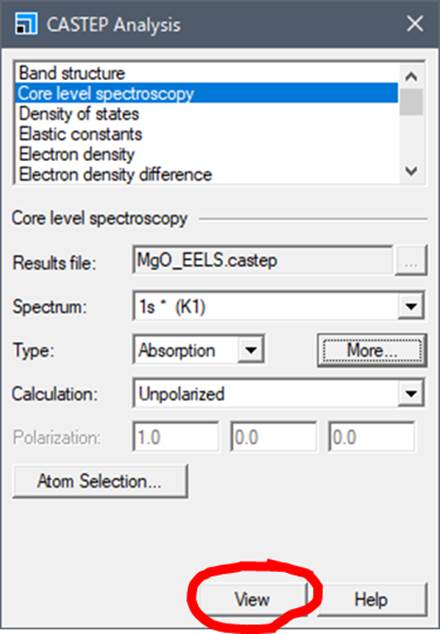
Press
"View" to display the O-K-edge ELNES/XANES of the selected oxygen
atom in the Materials Studio window. In this case, the following spectra were
obtained.
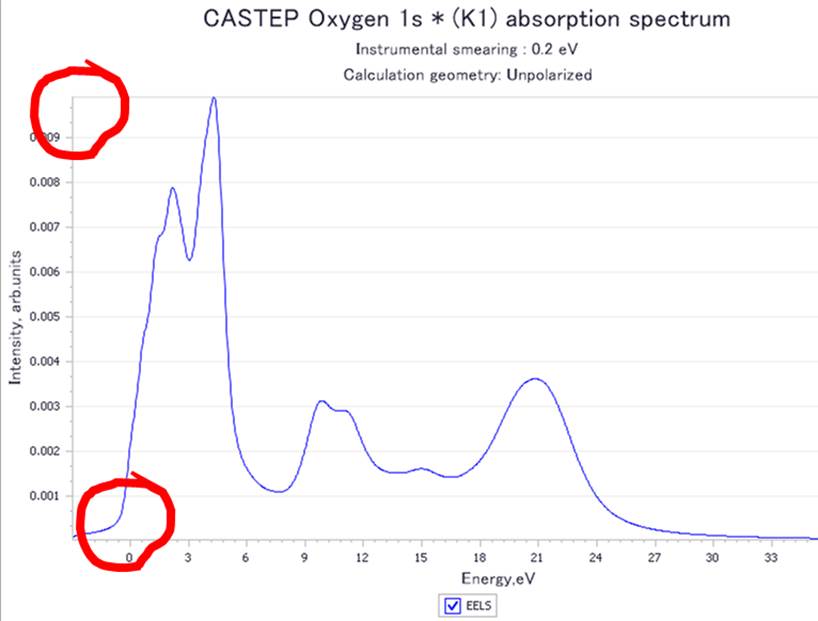
If you look at the horizontal axis, you can see that the peak
rises from zero. This indicates that the transitioned electrons are at the bottom
of the conduction band (the calculation is well done!!). Next, let's look at
the values at the top of the vertical axis. You can see that the value is not
normalized (non-one).
To get text file of this graph, please copy (Ctrl+c)
this.
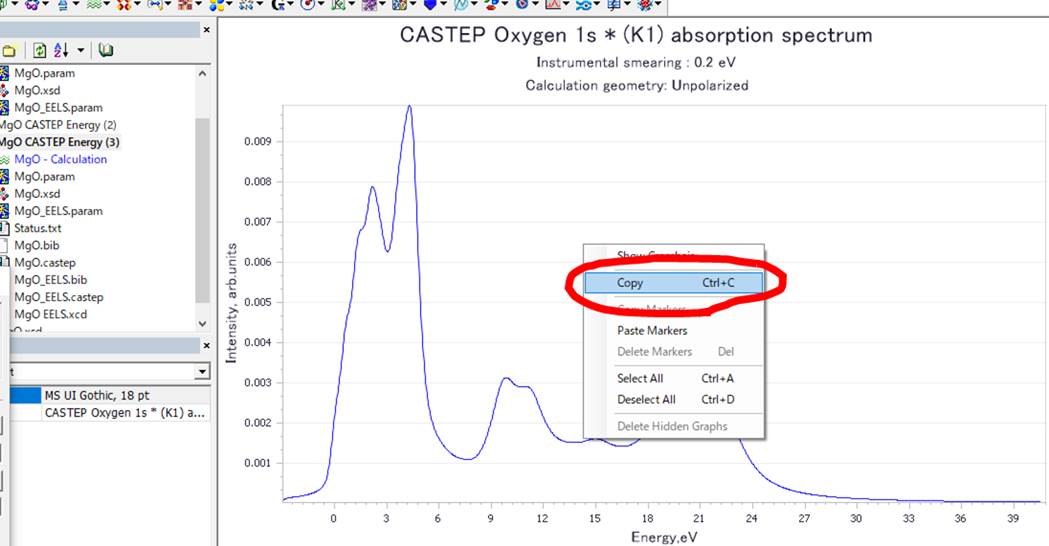
Then, you can paste (Ctrl+v) it to Excel or other text editor as below.
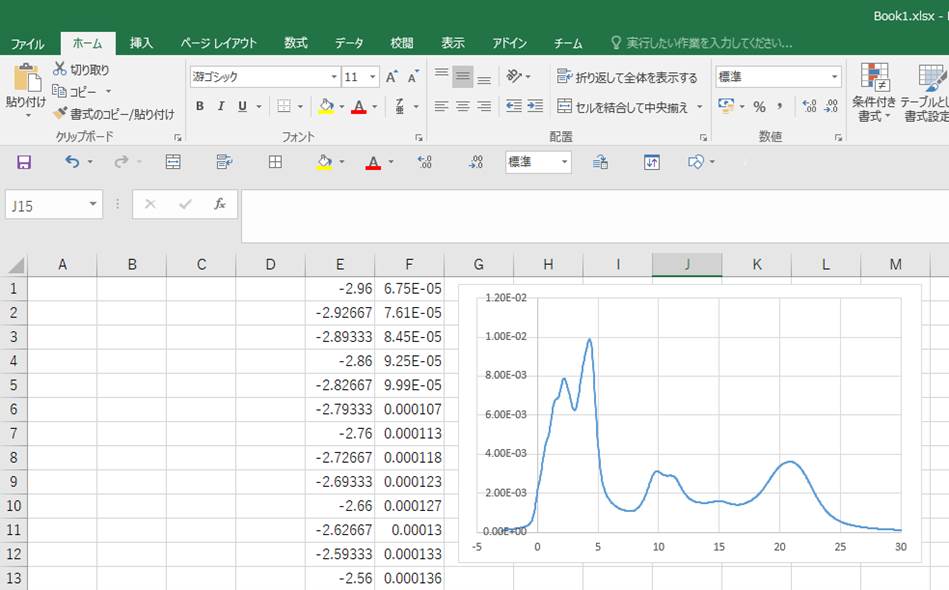
5.Calculate
Theoretical transition energy
Finally, lets calculate the
theoretical transition energy using the simulated data. Details are described in the article:
T. Mizoguchi et al. J. Phys.: Cond. Matter.,21 (2009)
104204-1-6.
First, you have
to simulate "Ground state" as below.
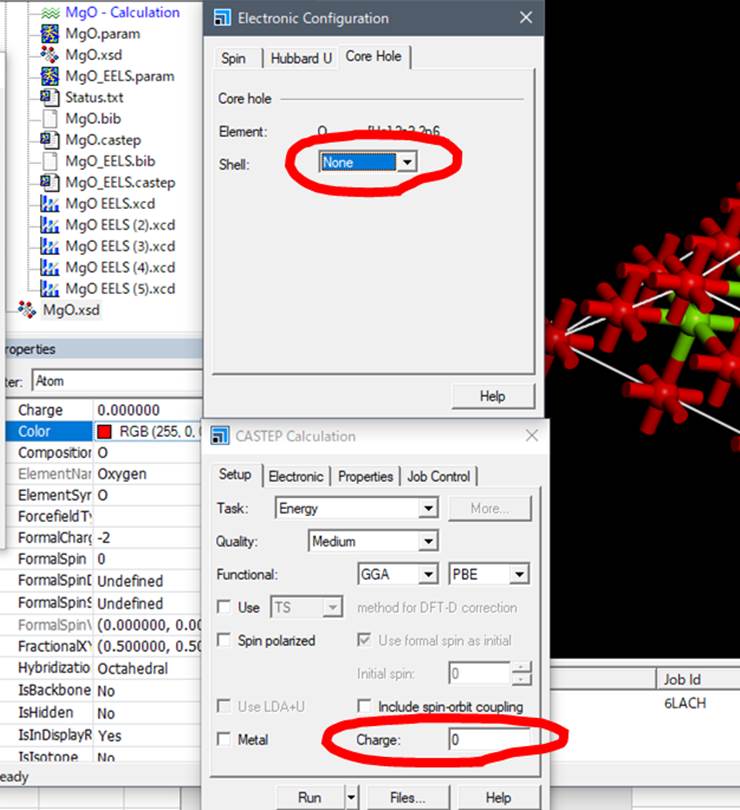
Actually,
you do not
need to simulate the spectrum. SCF simulation of the ground state is enough to calculate
the transition energy.
Now, you have performed two
simulations, that is Excited/Final state and Ground state. To obtain the
transition energy, you need following 6 values (which have been already
simulated).
1.Total
energy @ Ground state à this
value is around middle of case.castep.
2.Total
energy @ Final state à this
value is around middle of case.castep.
3.Single
atom total energy @ Ground state
à this value is around initial of case.castep.
4.Single
atom total energy @ Final state à this
value is around initial of case.castep.
5.Single
atom valence energy @ Ground state à this
value is around initial of case.castep.
6.Single
atom valence energy @ Final stateà this
value is around initial of case.castep.
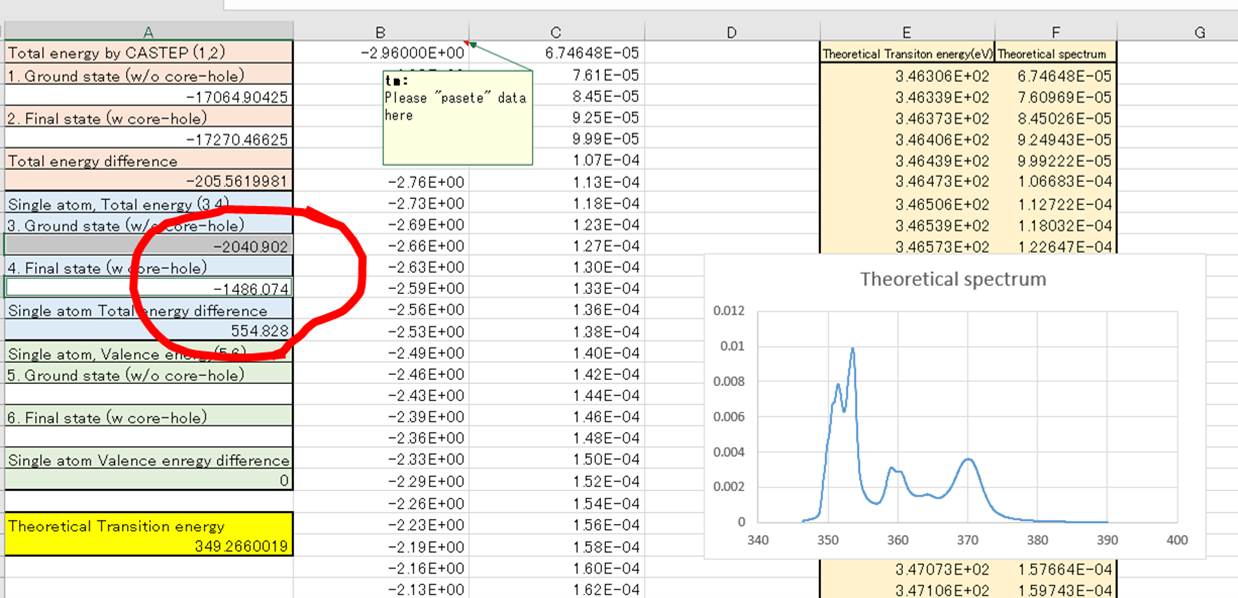
This is very simple math. For
convenience, you can use this Excel file (Here).The
file is as follows (sorry this is Japanese version Excel, but it must be used
in any Languages).
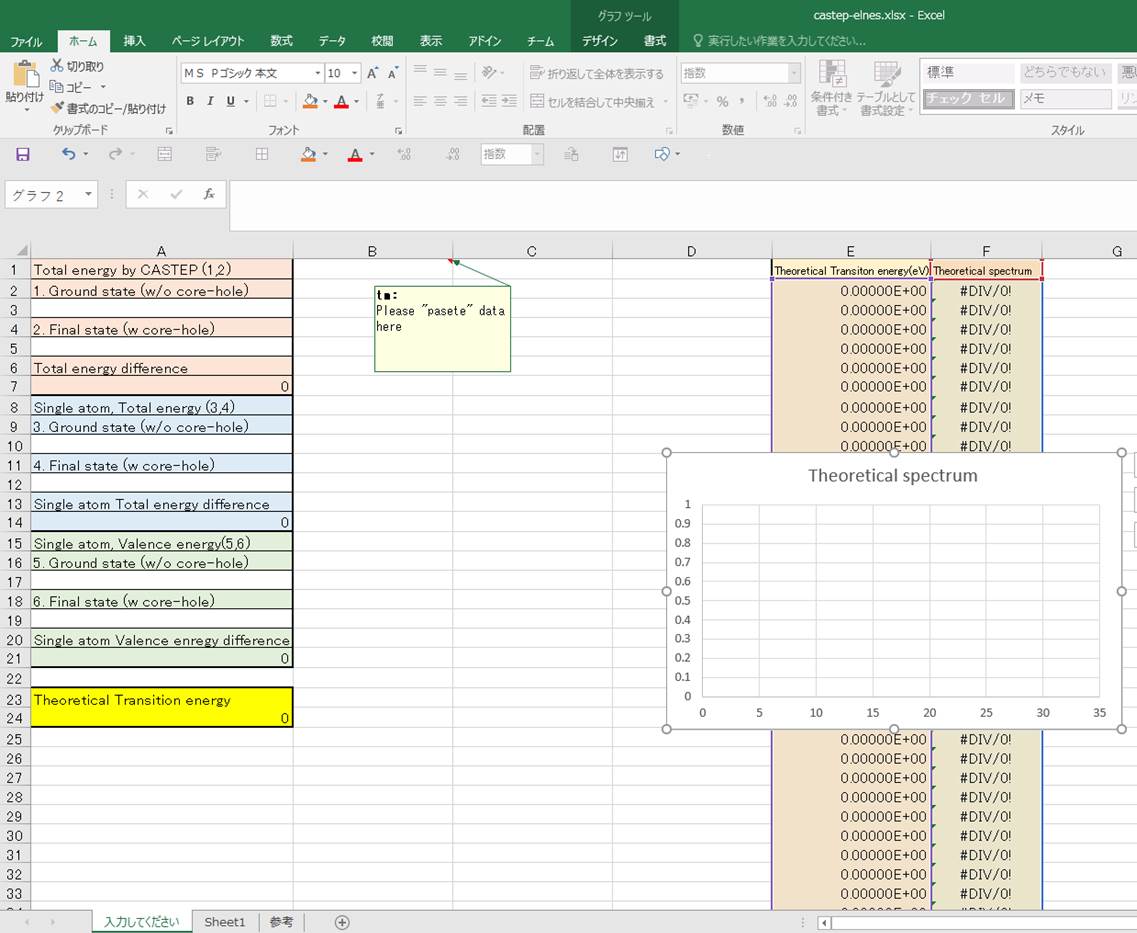
Column-E is "Theoretical
transition energy" (x-axis) and Columnn-F is "Theoretical
spectrum" (y-axis). The mathematics is as below :
(please see above reference for detail!!)
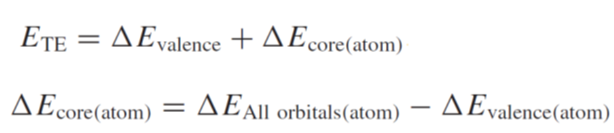
ETE is the
transition energy. By using above 1~6 values, you can obtain each values as below:
ΔEvalence=
2 - 1
ΔEcore(atom)=
(4 – 3) – (6 – 5)
*ΔEAll
orbitals (atom)= 4 – 3,ΔEvalence
(atom)= 6 - 5
First, lets
copy and paste the spectrum data from Materials Studio at B1-cell as below:
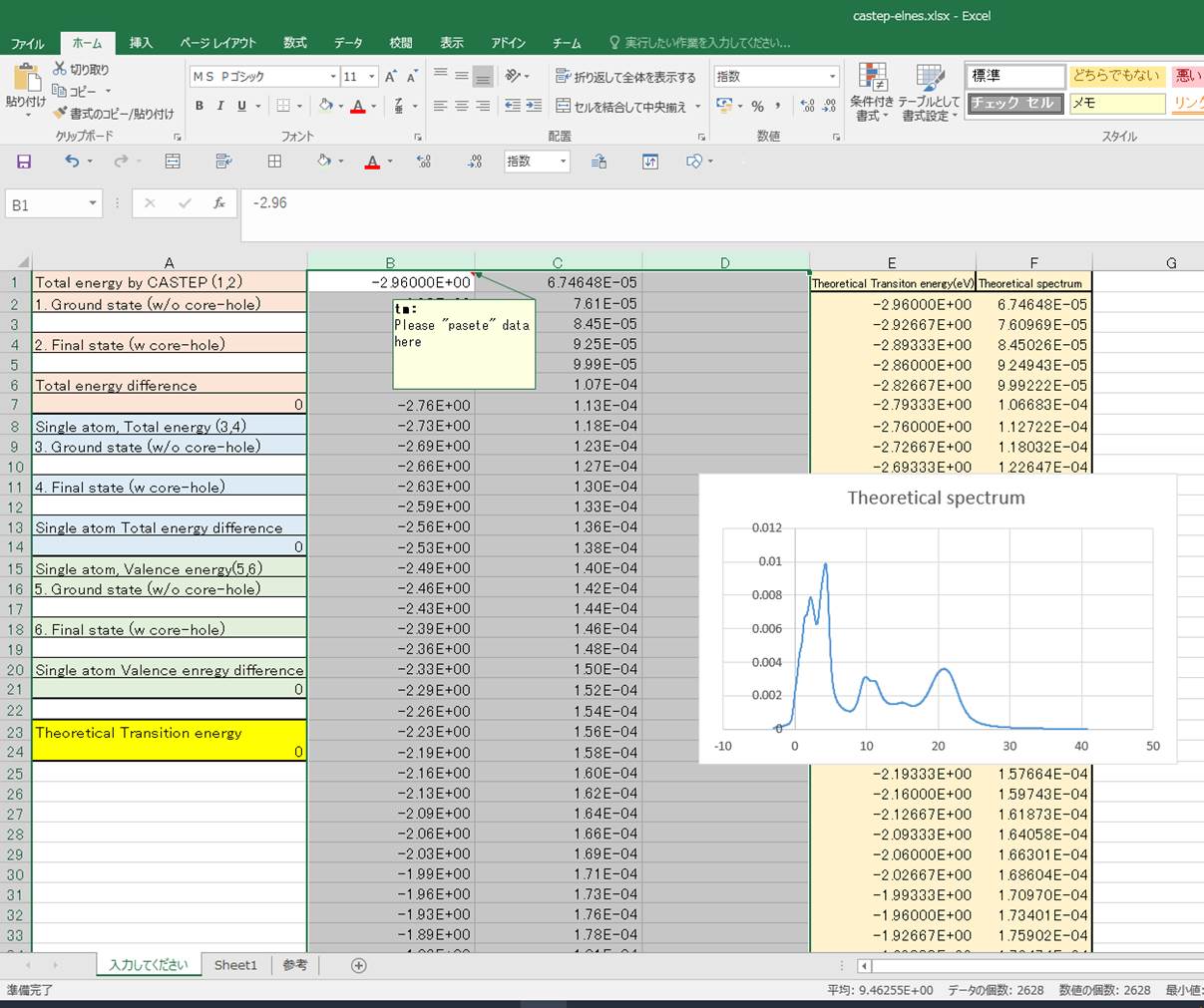
Then, lets input values in the
1st column.
1.Total
energy @ Ground state à this
value is around middle of case.castep.
2.Total
energy @ Final state à this
value is around middle of case.castep.
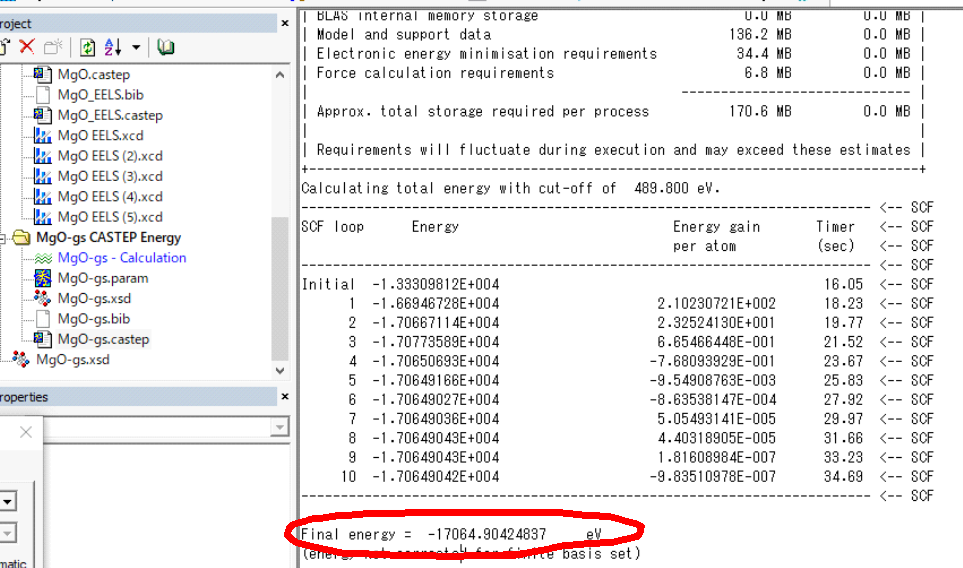
This "Final energy"
at Ground state and Final state are 1. and 2., respectively. Input these values
as below:
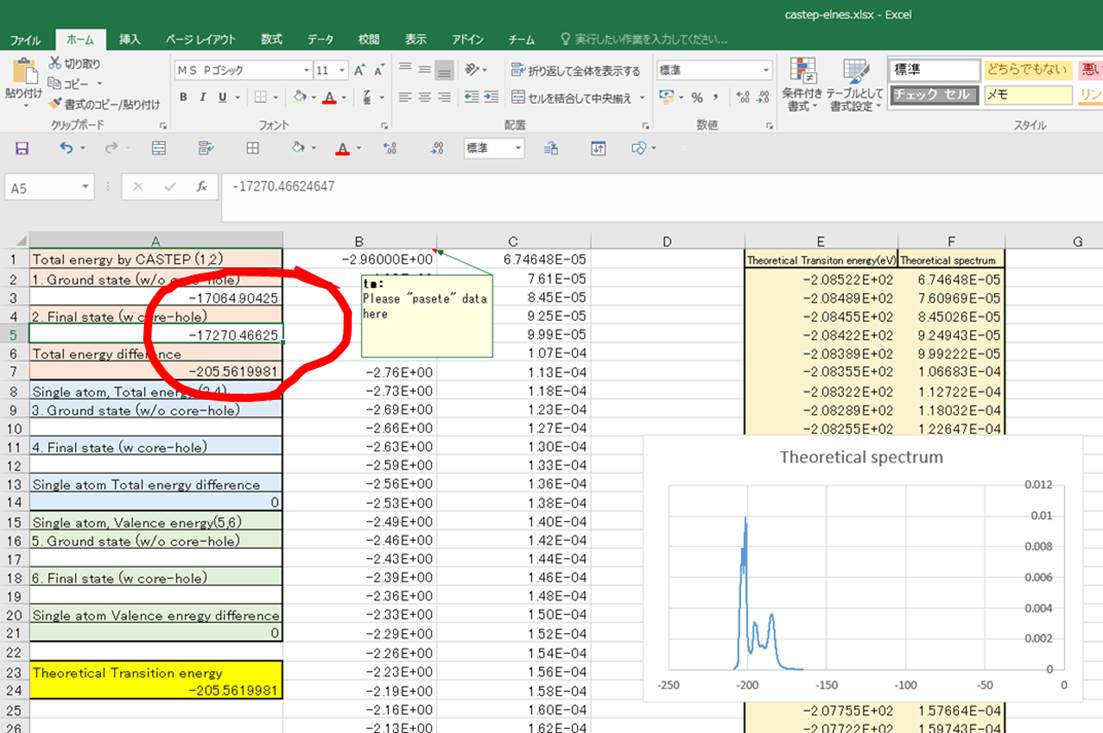
Then, lets obtain following
values:
3.Single
atom total energy @ Ground state
à this value is around initial of case.castep.
4.Single
atom total energy @ Final state à this
value is around initial of case.castep.
For instance, the value 3 is
given as below in case.castep
file.
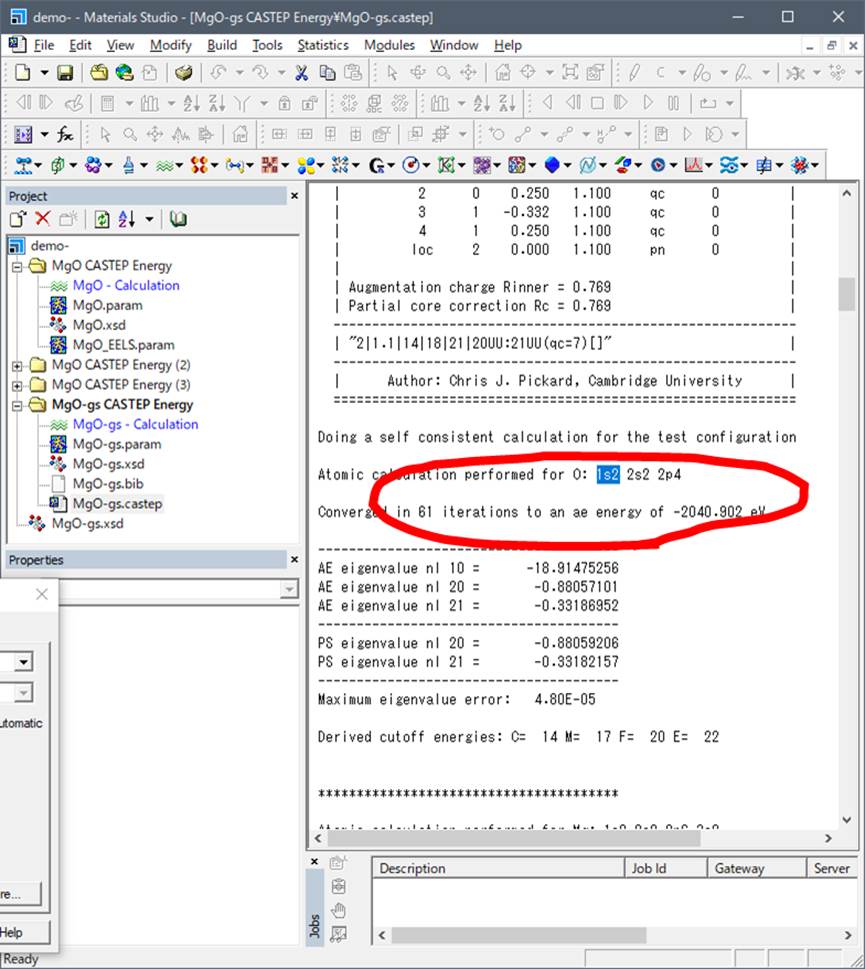
The
corresponding value at the final state is as below:
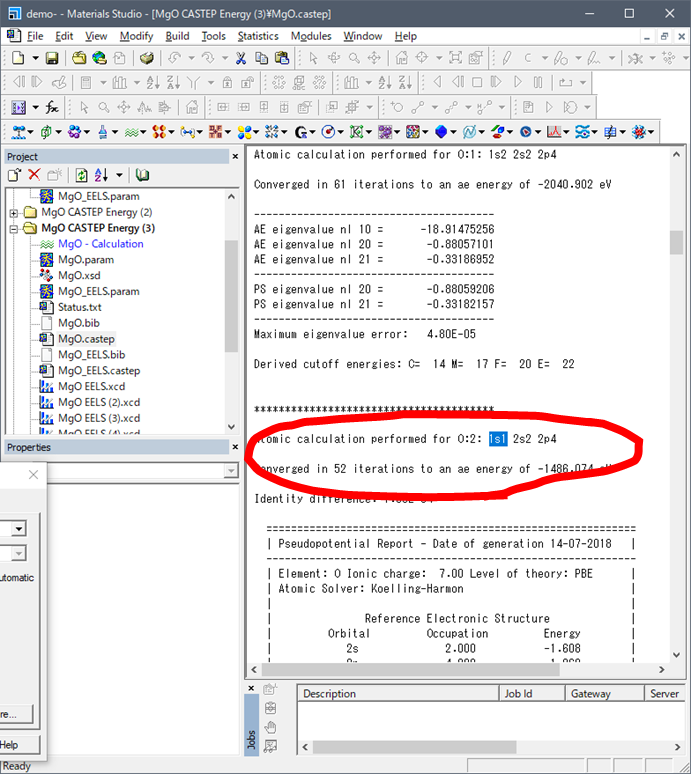
Now, you can see the number of electron at 1s orbital is one (1s1) at the Final state. Input
these values as values of 3. and 4. as below.
5.Single
atom valence energy @ Ground state à this
value is around initial of case.castep.
6.Single
atom valence energy @ Final stateà this
value is around initial of case.castep.
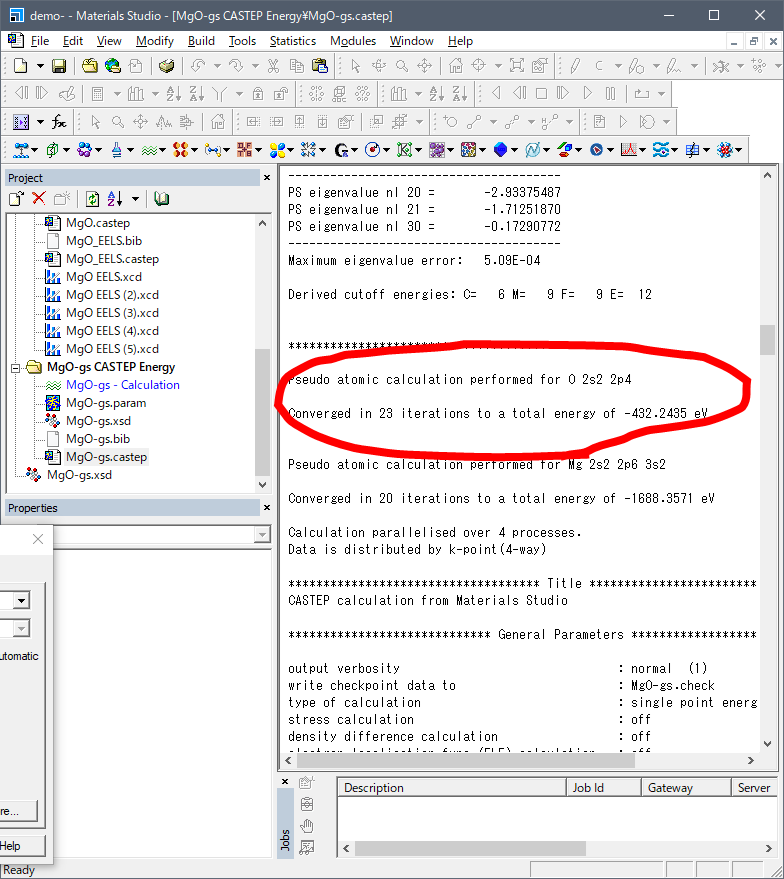
Next is 5 and 6. Please find "Pseudo
atomic calculation performed for" in your case.castep file.
For instance, you may find
following values. This is the valence energy of ground state.
The final state is as blow:
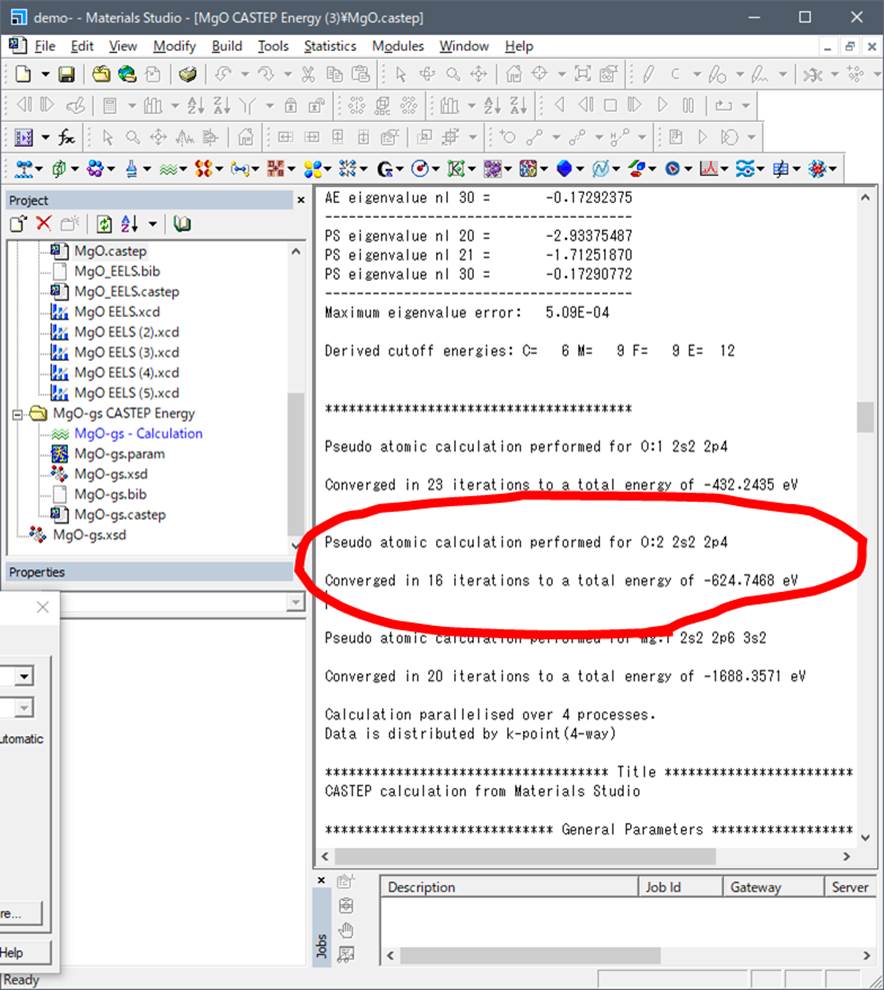
Be care that you have
introduced core-hole at O:2 (Not O:1!!), so you have to
use the valence energy of O:2. Input these values in 5 and 6.
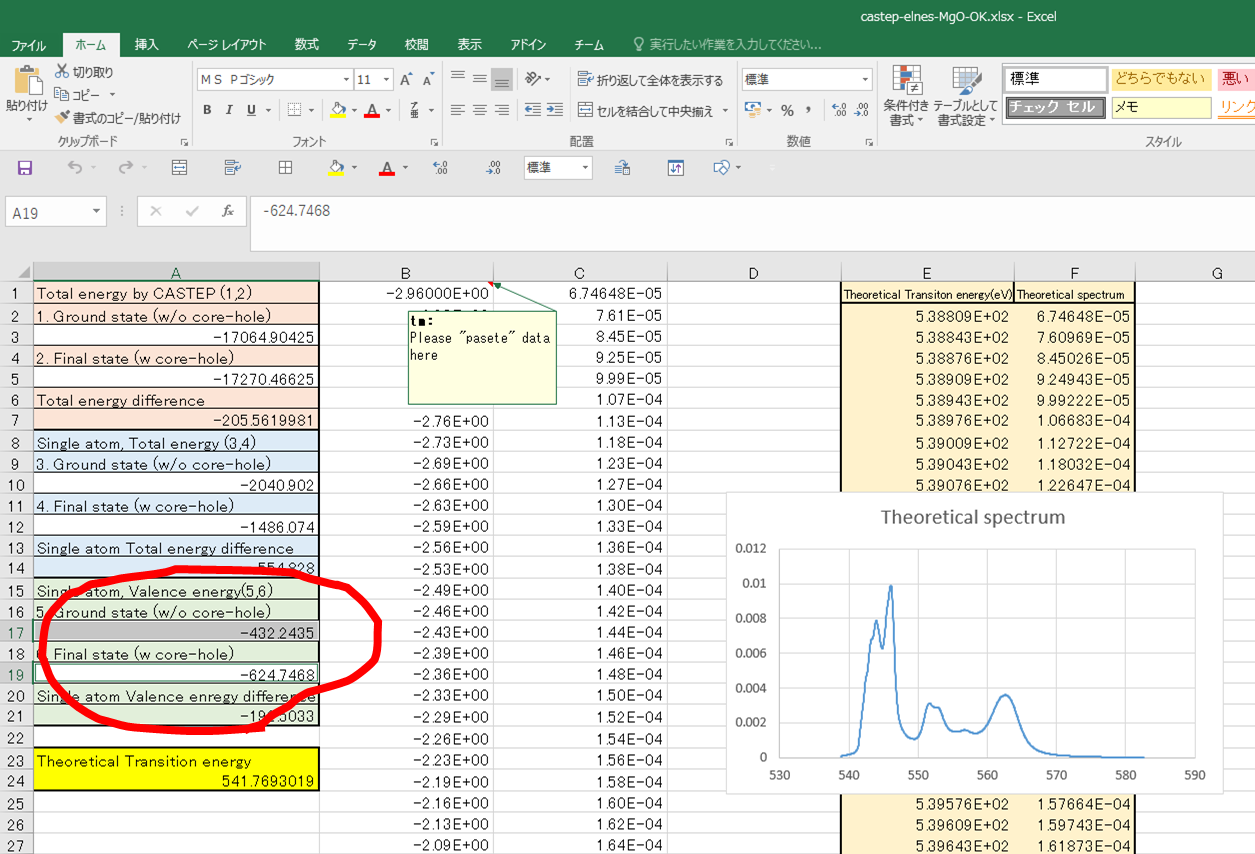
Congratulations! Now you got
the transition energy to be around 541.8eV!! This value is the threshold energy
of the calculated spectrum. You also obtain the theoretical spectrum with
theoretical transition energy.
Reference
CASTEP:
"First principles methods using CASTEP"
S. J. Clark, M. D. Segall, C. J.
Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne,
Z. Kristallogr., 220, 567–570
(2005).
CASTEP-ELNES/XANES:
"First principles calculation of spectral feature, chemical
shift, and absolute threshold of ELNES and XANES using plane wave
pseudopotential method"
T. Mizoguchi, I. Tanaka, S Gao, and C.J. Pickard,
J. Phys.: Cond. Matter.,21 (2009) 104204-1-6.
Liquid ELNES/XANES using CASTEP:
"An estimation of molecular dynamic behaviour
in a liquid using core-loss spectroscopy"
Y. Matsui, K. Seki, H. Hibara, T.
Mizoguchi
Scientific
Reports, 3 (2013) 3503-1-7. [free access]
Gas ELNES/XANES using CASTEP:
"Estimation of the molecular
vibration of gases using electron microscopy"
H. Katsukura,
T. Miyata, M. Shirai, H. Matsumoto, and T. Mizoguchi
Scientific Reports, 7 (2017), 16434-1-9. [free access]
First principles simulation of ELNES/XANES
using one-particle (DFT-GGA/LDA), two-particle (BSE), and multi-particle (CI)
methods:
"Basics and Applications of ELNES calculation"
(Invited Review)
H. Ikeno and T. Mizoguchi
Microscopy, 66 (2017) 305–327. [free access]
"Theoretical ELNES: one particle and many particle
calculations"(Invited Review)
T. Mizoguchi, W. Olovsson, H. Ikeno, and I. Tanaka
Acknowledgements:
Students
in my group, Prof. Hidekazu Ikeno@Osaka
Pef. Univ., Prof. Kobayashi@ Kyoto Inst. Tech., Mr. Saito@JEOL, Dr. A. Chatterjee@BIOVIA,
Prof. Chris J. Pickard @Cambridge University, Prof. SP. Gao@Fudan
University, and Prof. Isao Tanaka@Kyoto University.