CASTEPを用いたELNES/XANESの第一原理計算
東京大学・生産技術研究所・溝口照康
2014/9/15
ここでは第一原理擬ポテンシャル法であるCASTEPコードを用いたELNES/XANESの第一原理計算法を説明します.特に,アクセルリス社(現 BIOVIA)のMaterials
StudioというGUIを用いた手法について説明します.

ここでは酸化マグネシウム(MgO)のO-K端を計算してみます.
1.Materials Studio上でモデルを作製する.
File-> Importを行い,「Structures」のショートカットに行き,「Metal-oxides」の中のMgO.msiを呼び出します.
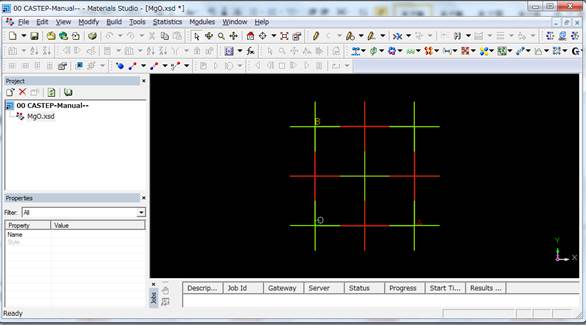
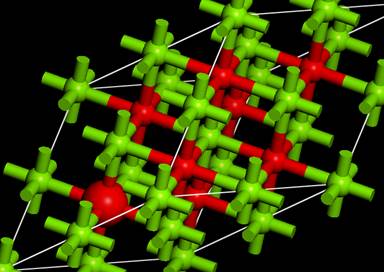
上図では酸素は赤色,Mgは緑色となっています.
上のようなunit
cellのままでも問題ありませんが,岩塩型構造はprimitive cellにできるので,primitive
cellにします.
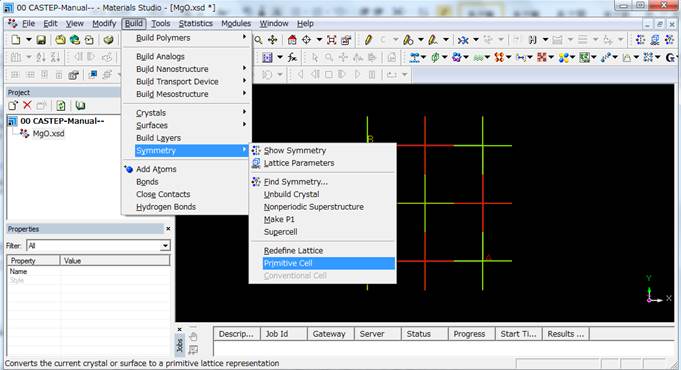
結果的にMg1O1の2原子のprimitive
cellができます.基底状態のバンド構造や状態密度(DOS)の場合はprimitive
cellを用いて,k点を増やした計算を行います.しかし,ELNES/XANES計算で導入する内殻空孔をprimitive
cellでは正確に計算できません.
たとえば酸素のK端を計算したい場合は,セルの中にある酸素の一つの1s軌道にホールを導入することになります.一方で,内殻空孔は電子状態に影響を及ぼします.バンド計算では計算するセルを無限に拡張する(周期的境界条件)ため,隣のセルの中にある内殻空孔を導入した酸素間に相互作用が発生します(下模式図).
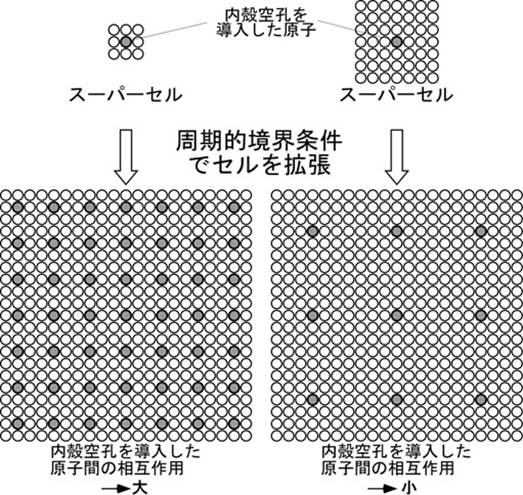
そこで,primitive
cellやunit
cellのような小さなセルではなく,それらを拡張したセル(スーパーセル)を用い,内殻空孔を導入した原子間の相互作用を小さくしまする必要があります(上図右側).そのために,MgOのprimitive
cellは2原子ですが,それを下のように拡張します.
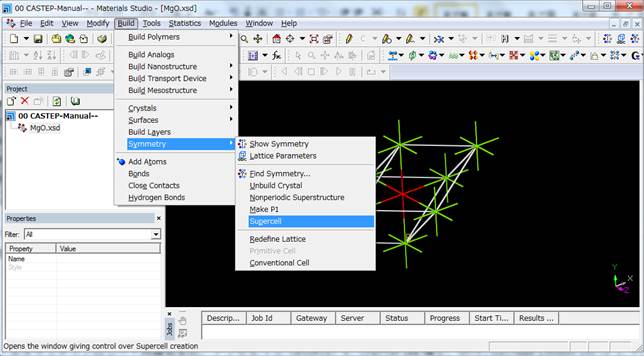
例えば2×2×2倍することで以下のような16原子のスーパーセルが作製されます.
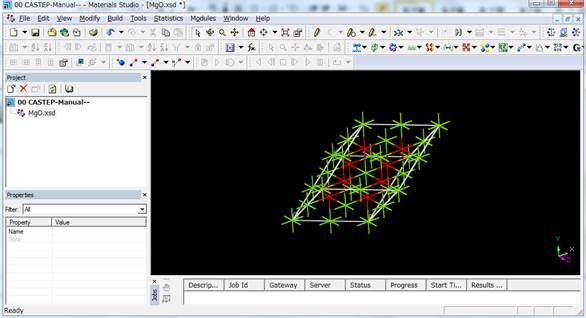
今回はこの16原子のスーパーセルを用います.また,このsupercellにした時点で結晶の対称性はP1(対称性なし)に落ちます.
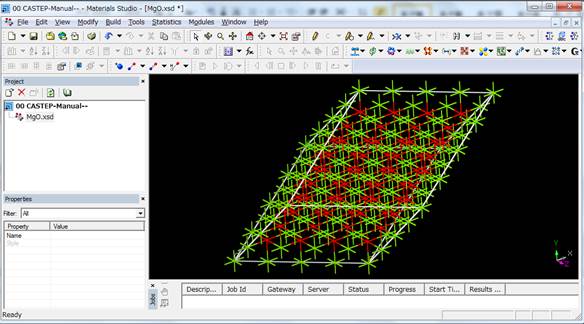
ただし内殻空孔を導入した原子間の相互作用を十分に小さくするためには10Å近く離す必要があることが分かっています.なので,MgOの場合はprimitive
cellを4×4×4倍した以下のような128原子のスーパーセルを用いるべきです.(128原子の計算時間は4コアで2,30分弱ほどです).
2.内殻空孔を導入する
今回は酸素のK端を計算するので,酸素を選びます.今時点ではスーパーセル内のどの酸素を選んでも問題ありません.
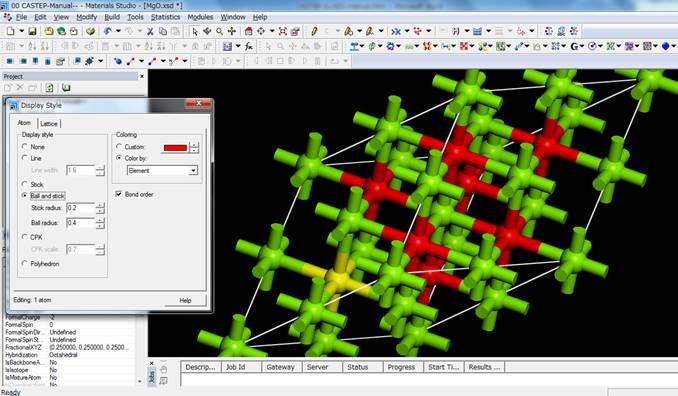
「Line」表示だと分かりにくいので,Display
styleでball
and stickにし,酸素を1つ選びます.ここでは上図左下の酸素(黄色)を選択してます.この酸素に内殻空孔を導入するために,選択したまま
Modify-> Electronic Configurationを選択します.
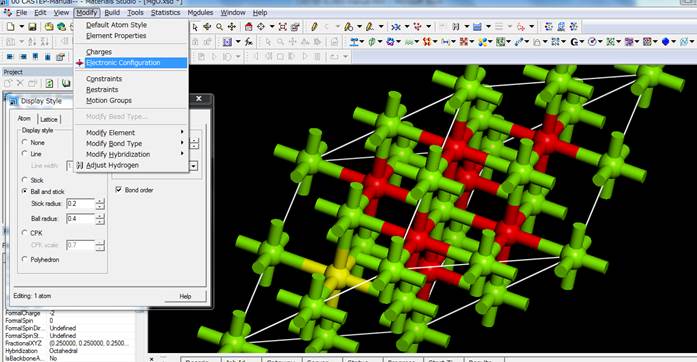
出てきたWindowの一番右のタブ「Core
hole」を選びます.
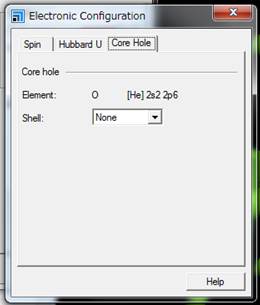
通常は基底状態なのでShellの欄がNoneとなってます.酸素のK端を計算するために1sにcore-holeを導入します.
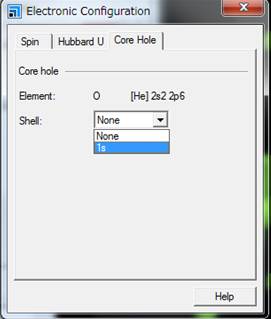
1sを選択したらwindowを閉じます.モデルをみても,見た目では内殻空孔が導入されているのか分かりません.確認するためには,Edit->Atom
Selectionを選択し,
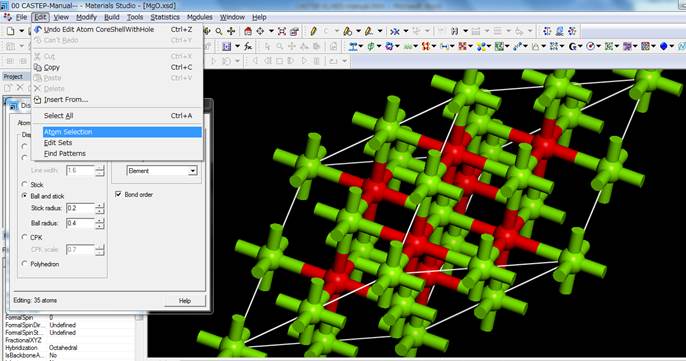
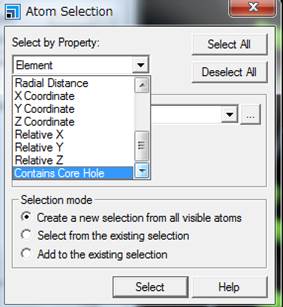
Contain
Core Holeを選択して,SelectするとCore
Holeを導入した原子が選択されます.わかりやすくするために,内殻空孔を導入した原子をDisplay
styleを使って,Ballのサイズを変えておくと便利です.
次に,Material
Studioはsupercellをつくると対称性がP1に落ちます.P1のままで計算するのは非効率なので,このセルに対称性を与えます.
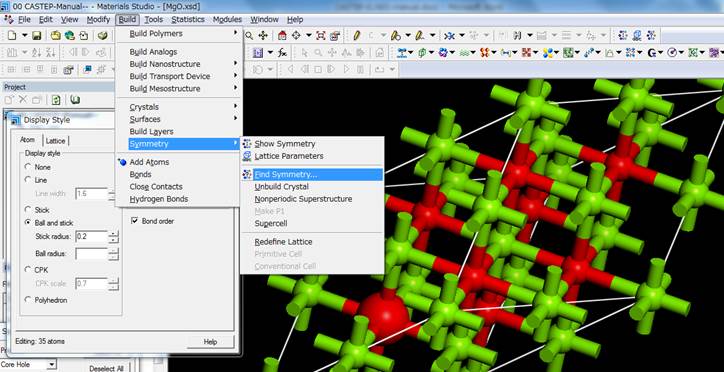
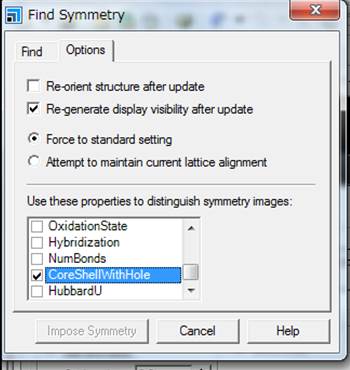
デフォルトの設定でsymmetryを探すと,元素の種類だけで対称性が検索されてしまいます.今回は内殻空孔を導入しており,内殻空孔を導入した励起状態の酸素と,内殻空孔を含まない基底状態の酸素とは区別すべきです.そのためにFind
symmetryのwindowの右のタブのOptionsを選択し,CoreShellWithHoleを選択しておきます.
次に,Findのタブにもどり,Find
symmetryをします.
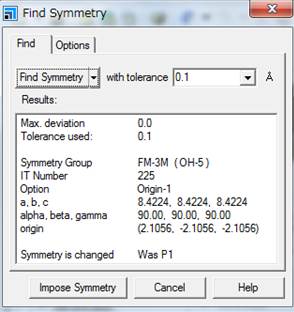
そうすると今回Fm-3mが見つかります.次にImpose
symmetryします.

その結果,上図のようなUnit
cell表現のスーパーセルが作製されます.Primitive cellの2×2×2の計算するために再びBuild->Symmetry->Primitive
Cellを実行します.
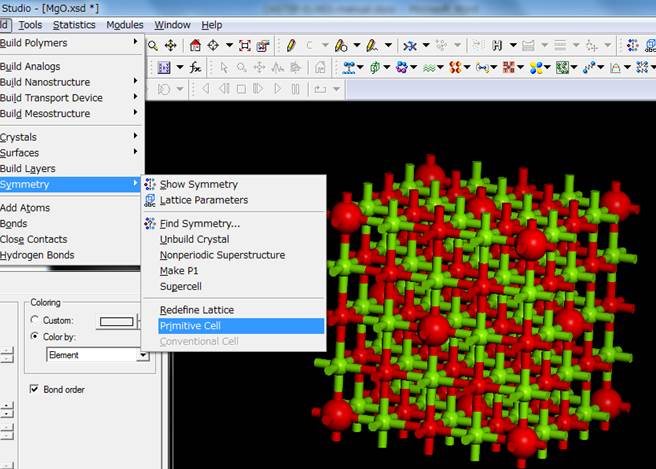
今回は以下のようなセルになりました.つまり,内殻空孔を導入した原子(大きな赤丸)がずれて,スーパーセルの端になっていることが分かります.これは,対称性を上げるために,内殻空孔を導入した原子を対称性の高いサイトに移動させたためです.
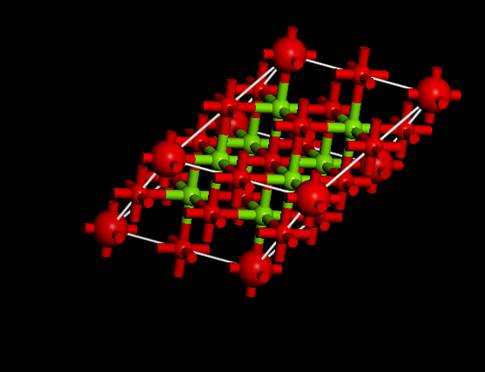
これで,計算を実行するためのセルが完成しました.
3.CASTEPを実行する
次にツールバーからCASTEPのアイコンを選択し,Calculationを選択します.
CASTEP
CalculationのWindowが立ち上がります.各タブを説明します.まずSet upタブについて,
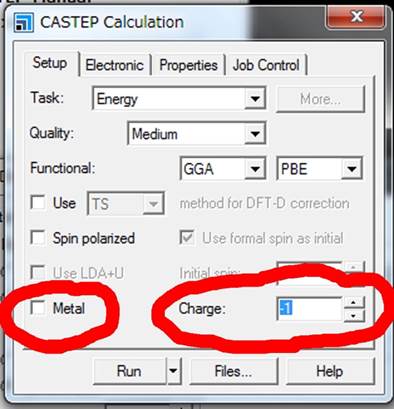
まずはMgOはMetalではないのでMetalのチェックを外します.次に重要なことですが,Chargeを-1にします.これは,Core
holeがスーパーセルにある場合にMaterial
Studioが勝手にセルのチャージを+1にするという“お節介”を正すためです.このお節介は,マイナス電荷の電子が内殻から抜けて,セルとしてプラスになったとMaterial
Studioが勝手に判断するためです.しかし,ELNES/XANESが反映しているのは内殻電子が伝導帯に入っている電子状態です.つまり,電子の位置が内殻軌道から伝導帯に移動しただけで,チャージとしてはニュートラルのはずです.これはMaterial Studio版CASTEPのみの問題で,著者がこれまで行ってきたすべての計算(Wien2k,OLCAO,ソース版CASTEP,DV-Xα等)ではすべてニュートラルなセルで計算しています.
そのようなMaterial
Studioの“お節介”を正すためにChargeを-1にしておきます.そうするとMaterial
Studioのお節介(+1)と入力した-1とが打ち消し合い,結果的にニュートラルな計算を行ってくれます.
次に,Electronicのタブに行きます.
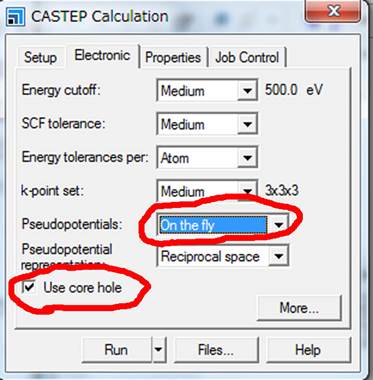
まず,左下の Use
core holeを選択します.つぎに,core-holeを導入するためにはPseudopotentialsの種類が On the
flyである必要があります.On the flyでは「その場で」pseudopotentialを作成するということです.On the
flyで内殻空孔を導入した擬ポテンシャル(励起擬ポテンシャル)を作製し,内殻空孔を導入すべき原子には励起擬ポテンシャルを適用し,他の原子には基底状態の擬ポテンシャルを適用します.
このパネルにEnergy
cutoffやk-pointsの設定もできます.ELNES/XANES計算の場合,Energy
cutoffやk-pointsは特に理論遷移エネルギーにきいてきます.いくつかの条件(たとえばMedium,
Fine,Ultra-fine)で計算して比較し,計算の精度を確かめる必要があります.
次にPropertiesタブに行きます.
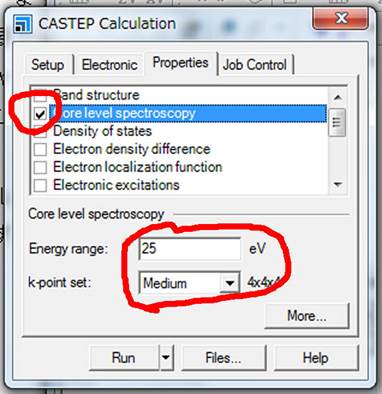
ELNES/XANESを計算するためにCore
level spectroscopyを選択します.下にEnergy rangeとk-point
setが出てきます.Energy
rangeは吸収端から何eV後ろまで計算するかということです.25eVの場合は吸収端から25eVぐらい後ろまでスペクトルを得ることができます.この値を大きくすると非常に計算時間がかかりますので,無駄に高エネルギーにしないほうが良いです.
k-point
setはスペクトル計算におけるk点です.Electronicのタブにもk-point
setがありましたが,Electronicタブの方は電子構造を計算する(SCF計算)するときのk点で,Propertiesタブの方は,SCF計算の結果をもとにスペクトルを計算するためのk点になり,k点が多いほど滑らかなスペクトルになります.今回はMediumで行います.
次にJob
Controlタブに行きます.
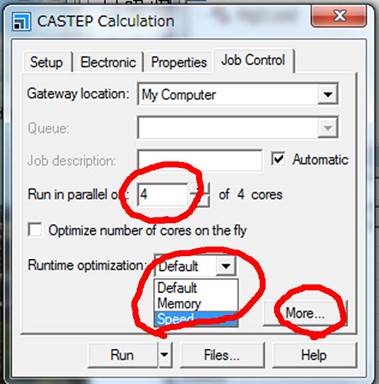
Run in
parallelは計算するPCのコア数になります.また,Runtime
optimizationがDefaultになってますが,これをSpeedにするとテンポラリーファイルをすべてメモリに格納してくれます.メモリを多く積んだPCであれば計算がかなり高速になります.次に,右下のMore...を選択します.そうすると以下のようなWindowがでます.
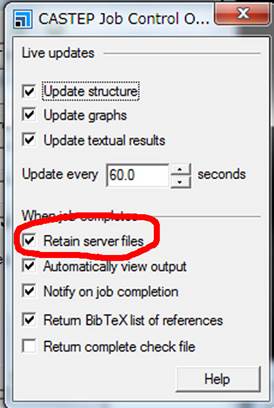
ここで,真ん中下ぐらいのRetain server filesを選択します.これは計算したファイルをすべてそのままにして,消さない,というオプションで,通常はOffになってます.これをonにする理由は,Material Studioが行う第二の“お節介”を回避するためです.その辺の説明は後にして,とりあえずこのままにして計算を実行します.元のWindowにもどり,Runを実行します.
4.ELNES/XANES計算結果をみる
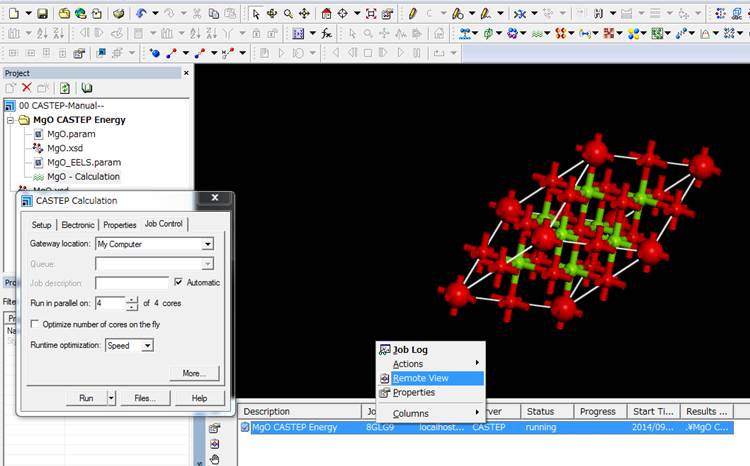
計算が走ると,下部のJobパネルに計算の状況が出てきます(Job
panelはView->Exploresで表示できる).Jobを上記のように選んで右クリックを押し,Remote
Viewを選択するとブラウザで以下のような計算ファイルのリストをみることができます.
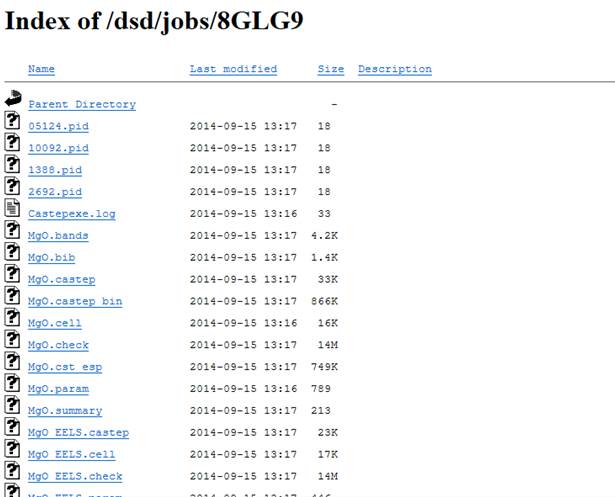
たとえば,MgO.castepが計算の主な出力ファイルで,現在どれくらいSCFサイクルが進んでいるのかがリアルタイムでわかります.
これらのファイルは実際にPCの中にできており,著者の場合は
C:\ProgramData\Accelrys\Materials
Studio\7.0\Gateway-x64\root_default\dsd\jobs\
の中に作成されてました.
しばらくすると計算が終了し,Materials
studioが以下のような表示になります.
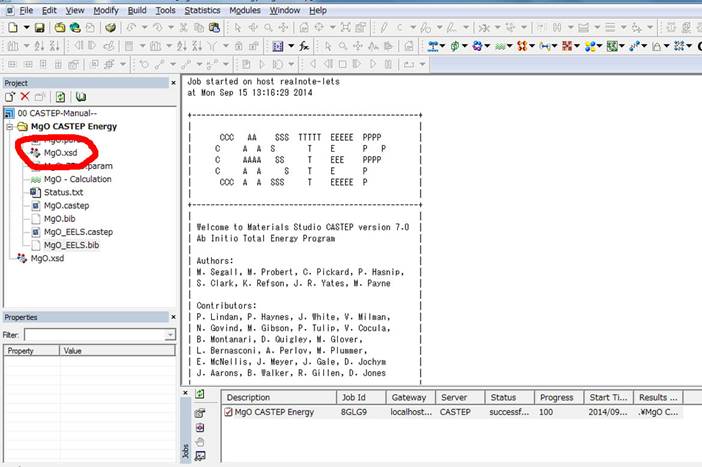
Corei5のPCで4coreでSCF計算が50秒(MgO.castepファイルの最後に出力されてます),ELNES/XANES計算が400秒ほど(MgO_EELS.castepの最後)ほどで終了しました.Retain
server filesが選択されているために下部のJobパネルで実行した計算が残ってます.次に計算したELNES/XANESを表示させるために,左のProjectの計算したフォルダのしたの計算モデル(上図左上の赤丸)をダブルクリックして選択します.
次にCastepボタンのAnalysisを選びます(下図).
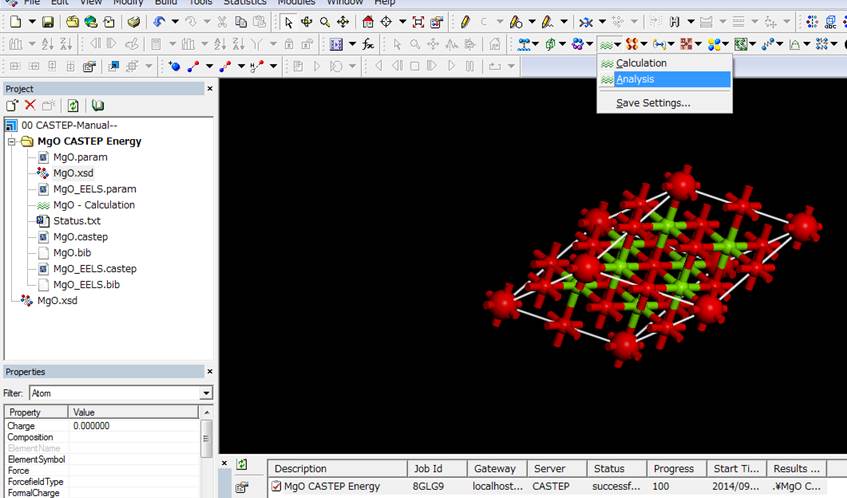
次に,内殻空孔を導入した酸素(大きな赤い酸素)を選択し,CASTEP
AnalysisのWindowでCore
level spectroscopyを選択します.
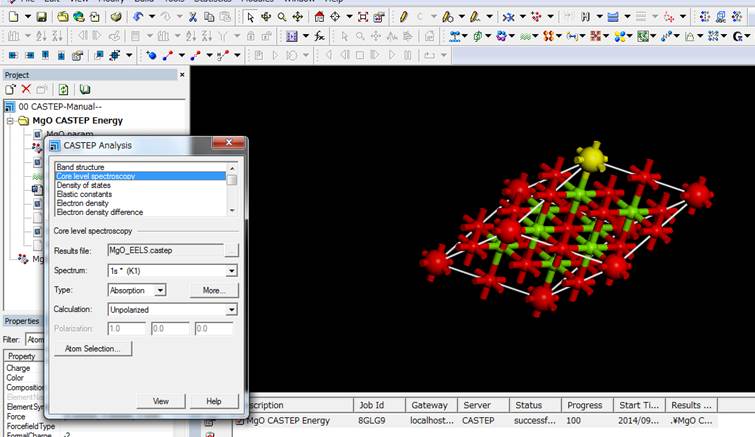
Core
level spectroscopyは選択した原子のELNES/XANESを表示してくれます.TypeでAbsorptionとEmissionが選択でき,Calculationで,UnpolarizedとPolarizedを選択することができます.ELNES/XANESはAbsorptionです.また注目する原子の電子構造に異方性がある場合,スペクトルに方位依存性があらわれます.異方性がある場合は,Polarizedにして,100や010,001にしてViewすると各方位に対するスペクトルを得ることができます.MgOの場合,酸素もMgにも異方性は無いのでUnpolarizedで行います.
また,Typeの右のほうにあるMoreを選択すると以下のようなWindowが立ち上がります.
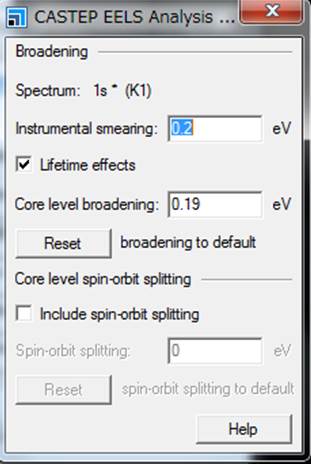
上部のInstrumental smearing,Core
level broadeningを変えることで計算のブロードニングを変えることができる.Instrumental
smearingがGaussian関数,Core
level broadeningがLorentzianであり,二つとも指定している場合はVoigt関数になります.今回はデフォルトの設定のままViewを選択します.
そうすると以下のようなスペクトルが得られました.
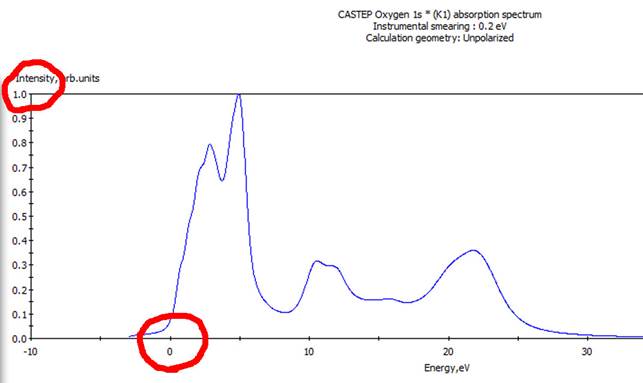
ここで注目するところの一つが横軸で,ゼロからピークが立ち上がっていることが分かります.これは遷移した電子が伝導帯下端にいることを示してます(計算がきちんとできているということです).次に縦軸上(図左上)の値をみてみます.1.0になっていることが分かります.これがMaterial Studioの二つ目の“お節介”で,Material
Studioでは計算したスペクトルをすべてノーマライズしてしまってます.このことは,結晶内に異なる原子サイトがある場合に問題が出てきます.結晶内に同種の元素で結晶学的に異なるサイトが複数個ある場合,各サイトでスペクトルを計算し,足し合わせる必要があります.その際にスペクトル強度をノーマライズしてしまうと正確に足し合わせることができなくなります.
そこで,サーバーに残しておいた計算ファイルが役に立ちます.
著者のPCでは
C:\ProgramData\Accelrys\Materials
Studio\7.0\Gateway-x64\root_default\dsd\jobs\
下にデータが残されてます.
フォルダの中身は以下のようになってます.
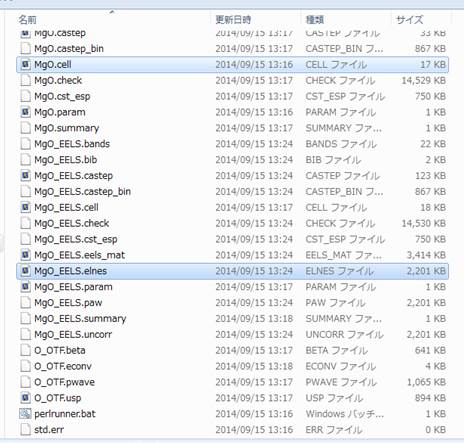
ここで,MgO.cellが計算の入力ファイルで,結晶構造やk点,On-the-fly用Stringなどの情報が含まれてます(ちなみにMgO.paramが計算の条件ファイルでcut off
energy等の条件が書かれてます).中身をテキストエディターなどであけると以下のようになってます.

一番上には結晶格子に関する3×3の行列があり,ここから格子定数(a, b, c,
α,β,γ)が分かります.中ごろには各サイトの位置と元素種に関する情報があります.ここで,酸素(O)がO:1とO:2に分かれていることが分かります.これは,酸素の一つが内殻空孔を含む“励起状態酸素”であるのに対し,残りの酸素が“基底状態酸素”になります.励起状態酸素は一個のはずなので,O:1がそれにあたることが分かります.
さらに,MgO_EELS.elnesがELNES/XANESの計算結果のテキストデータです.中身をみると以下のようになってます.
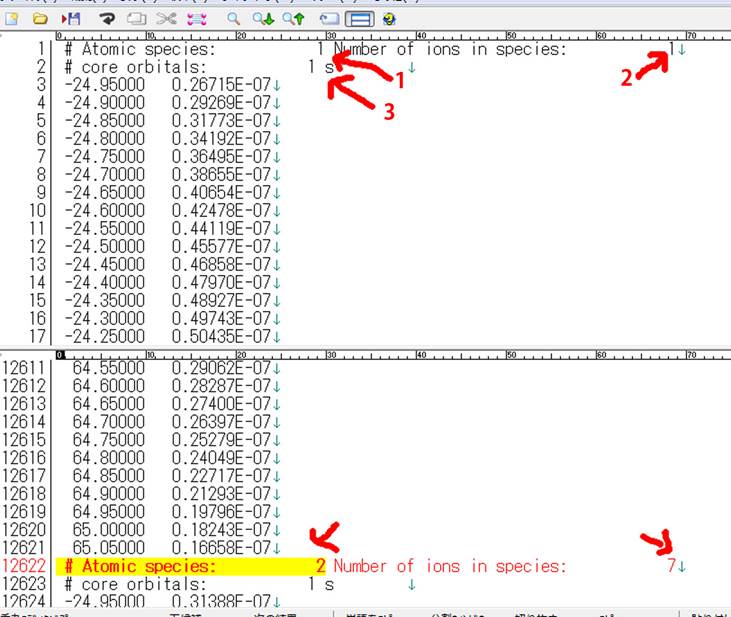
上図の上部に1,2,3とありますが,それぞれ
1.#Atomic species: 元素種のことで,上述のMgO.cellから今回3種類(O:1,
O:2, Mg:1)が存在しており,その一番目つまり,O:1ということ
2.Number of ions in species:1.の元素種の中の何番目かを示しており,O:1に属する酸素は一個のみ.O:2は7こあるので,7個出てくる.上図の下部では#Atomic
species: 2で Number of ions in species: 7の部分を示している.
3.# Core orbitals:その元素種の内殻の種類.O:1では1sに内殻空孔を導入して,内殻は1sのみしかないので“1s”しか出ないのでシンプル.しかし,Mgになると1s, 2s,
2px, 2py, 2pzが出力されている.
今回O:1の1s軌道にしか内殻空孔を導入してないので,他のO:2やMg:1はすべて無視して,
1.#Atomic species:=1
2.Number of ions in species:=1
3.# Core orbitals:1s
の部分だけをとりだし(1803行ほど),Excelなどでよみこみます.一列目がエネルギーで,二列目がスペクトルになります.Unpolarizedだと二列ですが,Polarizedの場合は3列(x, y, z)出てきます.
表示させると以下のように得られます.
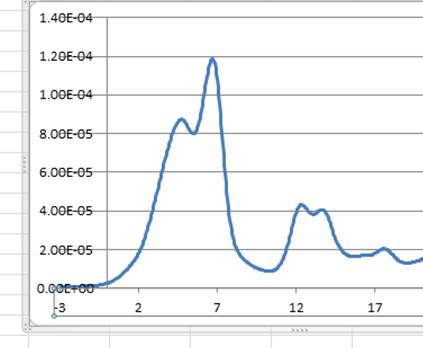
縦軸が規格化されてないことが分かります.
5.遷移エネルギーの計算方法
最後にCASTEPを用いた遷移エネルギーの算出方法を説明します.詳細は論文T. Mizoguchi et al. J.
Phys.: Cond. Matter.,21 (2009) 104204-1-6.をご参考ください.まず,ここまでに内殻空孔を含んだ遷移状態の計算を行ってきました.遷移エネルギーの計算では,遷移状態のエネルギーと基底状態のエネルギーの差を計算します.そのために,基底状態の計算をする必要があります.先ほどのMgO 16原子のモデルで,内殻空孔を入れましたが,その内殻空孔を戻します.

上では,MgO.xsdであったものをMgO-gs.xsdに変えてます.このままCASTEPの計算を実行しますが,なるべく同じ条件で計算します.
SetupパネルのChargeを0にもどし,ElectronicパネルのUse core
holeを外します.計算を実行すると以下のようなスペクトルが得られます.これが基底状態におけるスペクトルになります.

(*遷移エネルギーの計算のためには基底状態のエネルギーだけあればよいので,基底状態のスペクトルを出す必要はありません)
ここで,遷移エネルギーを計算しますが,必要な値は
1.基底状態の全エネルギー(castepファイル中ごろ)
2.遷移状態の全エネルギー(castepファイル中ごろ)
3.基底状態の単原子の全エネルギー(castepファイル初めあたり)
4.遷移状態の単原子の全エネルギー(castepファイル初めあたり)
5.基底状態の単原子の価電子エネルギー(castepファイル初めあたり)
6.遷移状態の単原子の価電子エネルギー(castepファイル初めあたり)
の6つです.
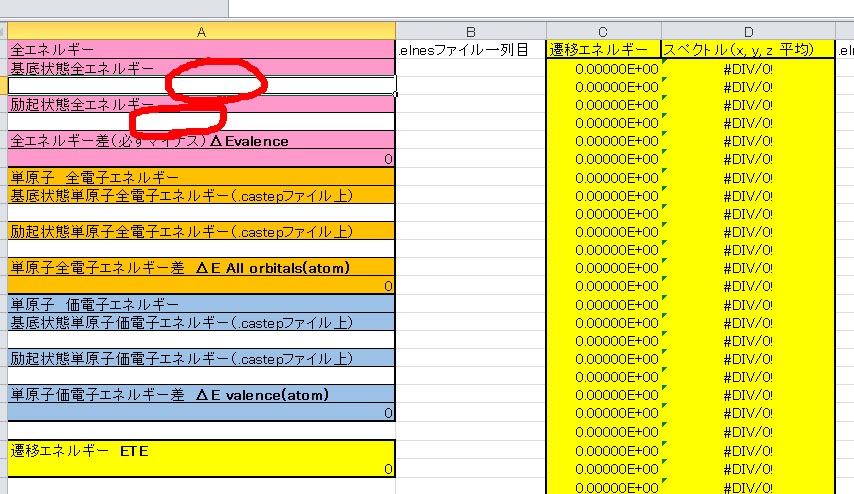
計算するのに便利なエクセルファイルを公開しております(こちら).まずダウンロードしてください.エクセルでそのまま開くと以下のようなものになってます.
カラフルですが,A列に上にあげた1〜6の値を入れます.B列とE〜G列は.elnesファイル(今回はMgO_EELS.elnesファイル)の各列を入力します.それでは各欄に値を入れてみます.
1.基底状態の全エネルギー(castepファイル中ごろ)
2.遷移状態の全エネルギー(castepファイル中ごろ)
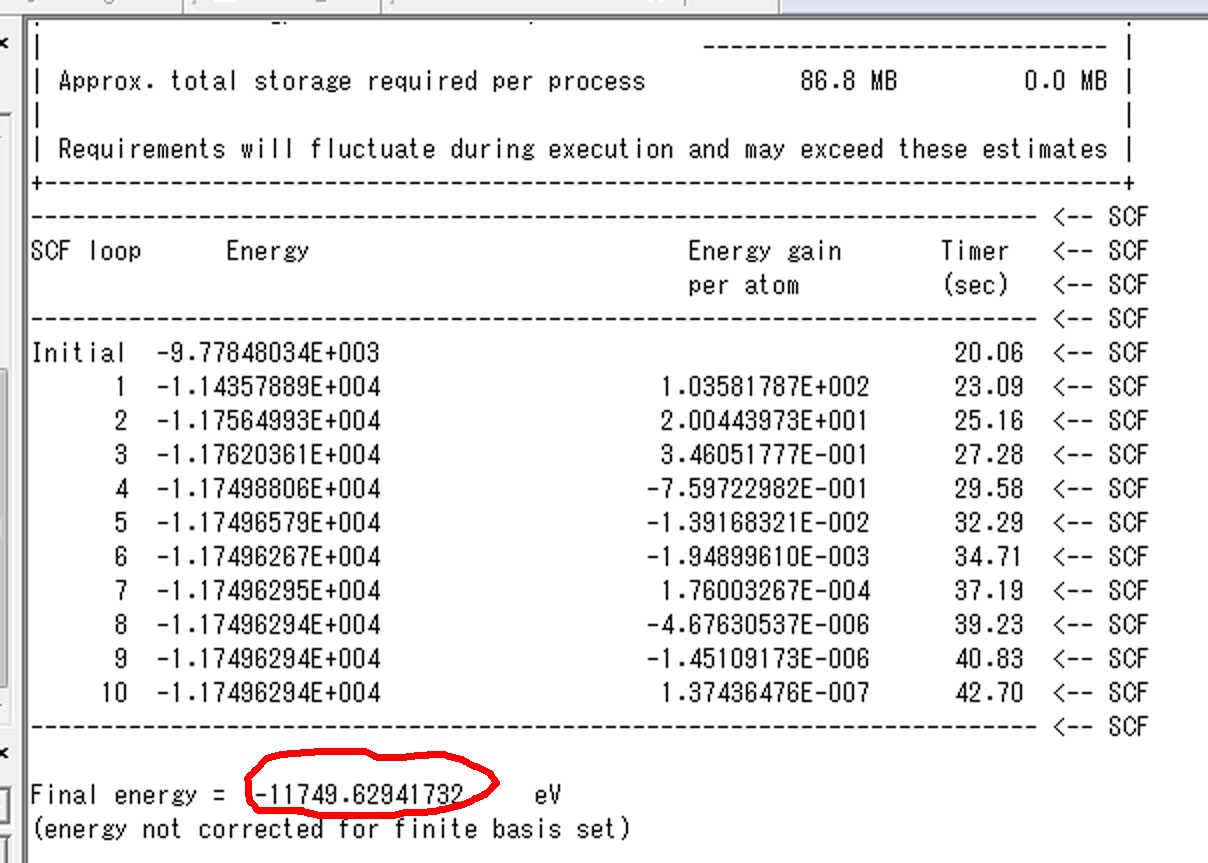
基底状態のMgO.castepの中ごろに以下のようにSCFでの全エネルギー―の値が出てます.
このFinal
energyが基底状態の全エネルギーとなります.同様に励起状態の.castepファイルから励起状態の全エネルギーが分かります.二つの値をエクセルの以下の欄にいれます.
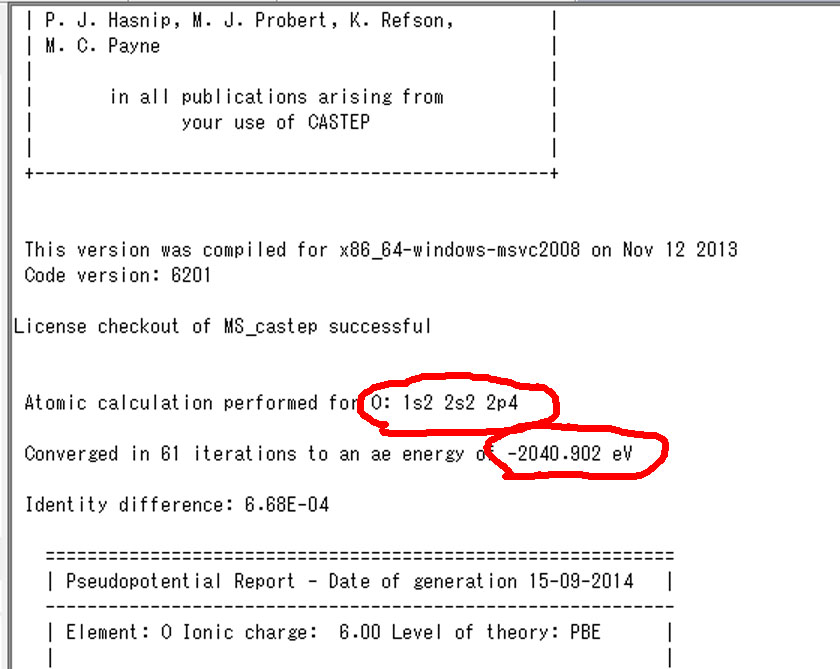
3.基底状態の単原子の全電子エネルギー(castepファイル初めあたり)
4.遷移状態の単原子の全電子エネルギー(castepファイル初めあたり)
次に,基底状態および遷移状態のMgO.castepファイルの上の方に,単原子の全電子エネルギーが出力されてます.
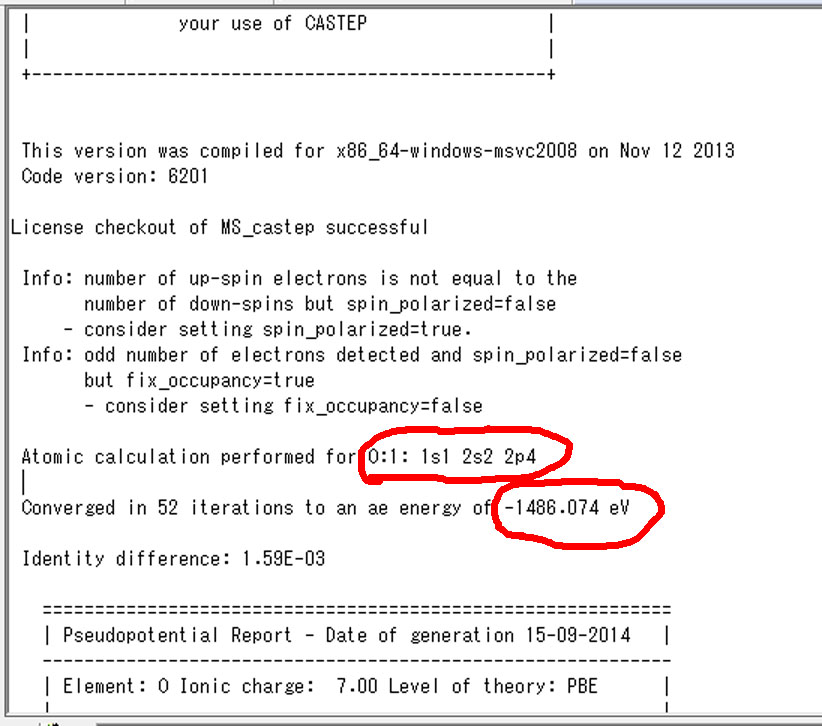

上図で左が基底状態,右図が励起状態です.励起状態の結果から一番目の酸素の1s軌道の占有電子数が一つ減ってることが分かります.なので,基底状態でも一番目の酸素の値を使用します.この二つの値をエクセルの単原子の全エネルギーの欄に入力します.
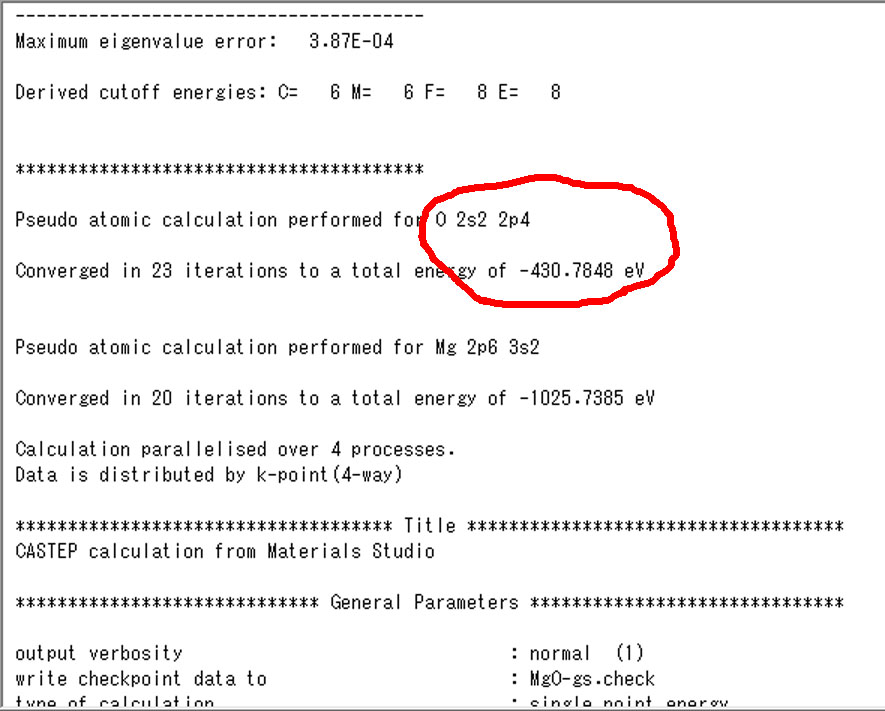
5.基底状態の単原子の価電子エネルギー(castepファイル初めあたり)
6.遷移状態の単原子の価電子エネルギー(castepファイル初めあたり)
次が単原子の価電子エネルギーです.これも.castepの初めの以下のあたりに出力されてます.
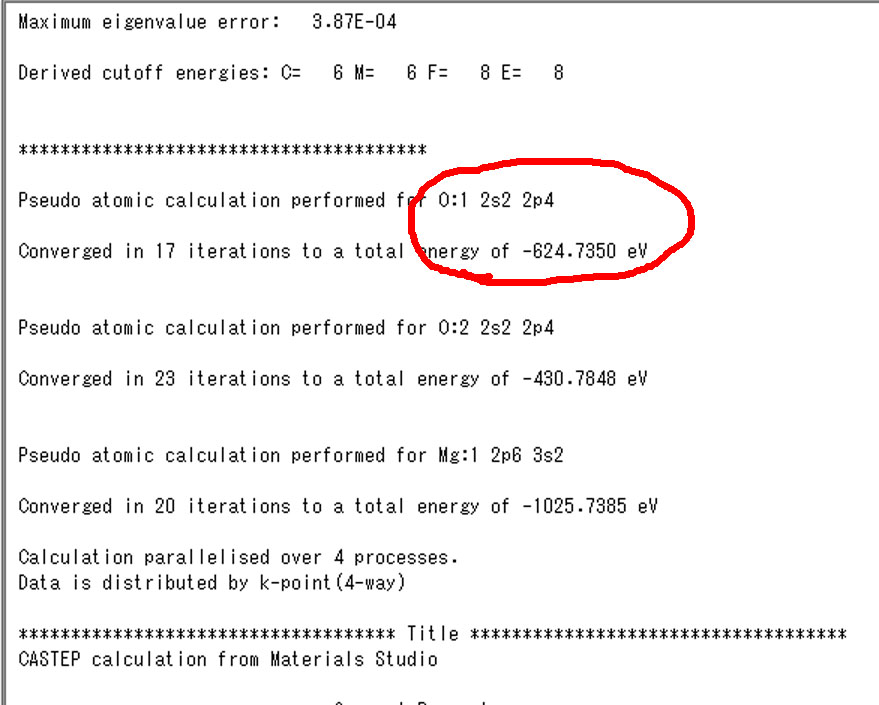
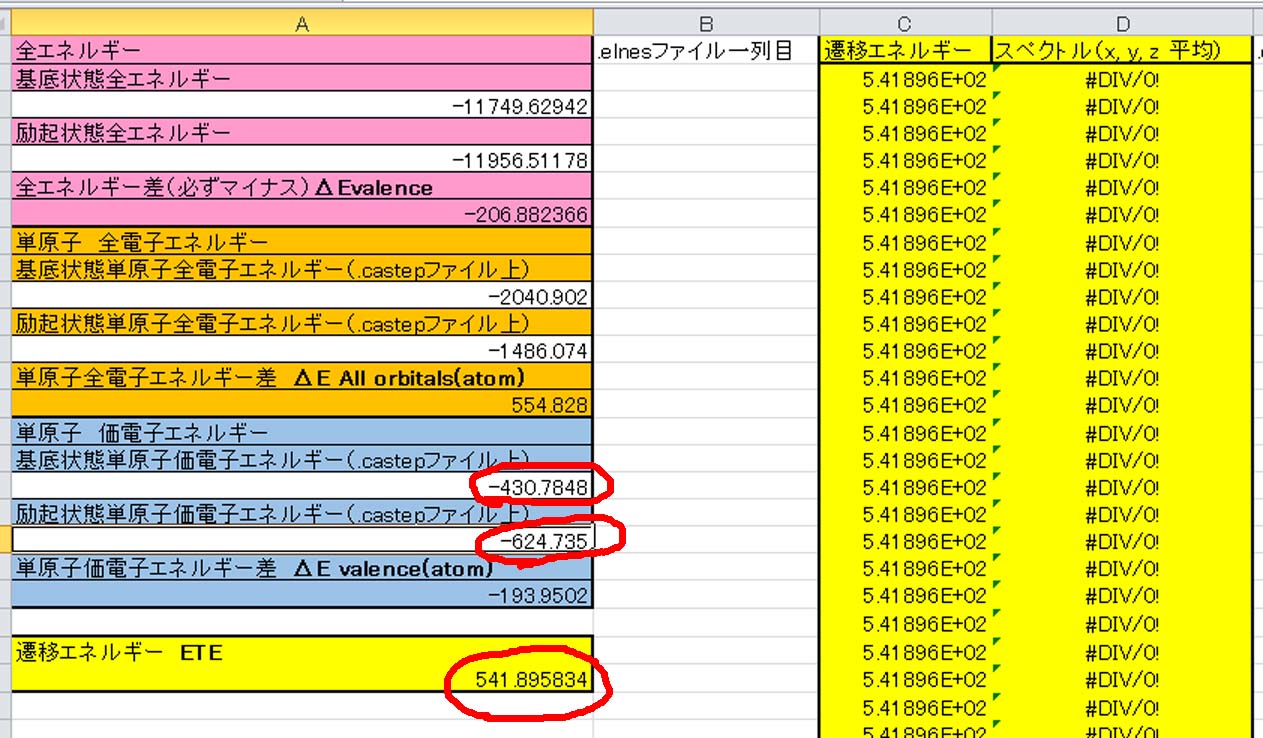
左側が基底状態で,右が励起状態です.これらの値をエクセルファイルに入力します.
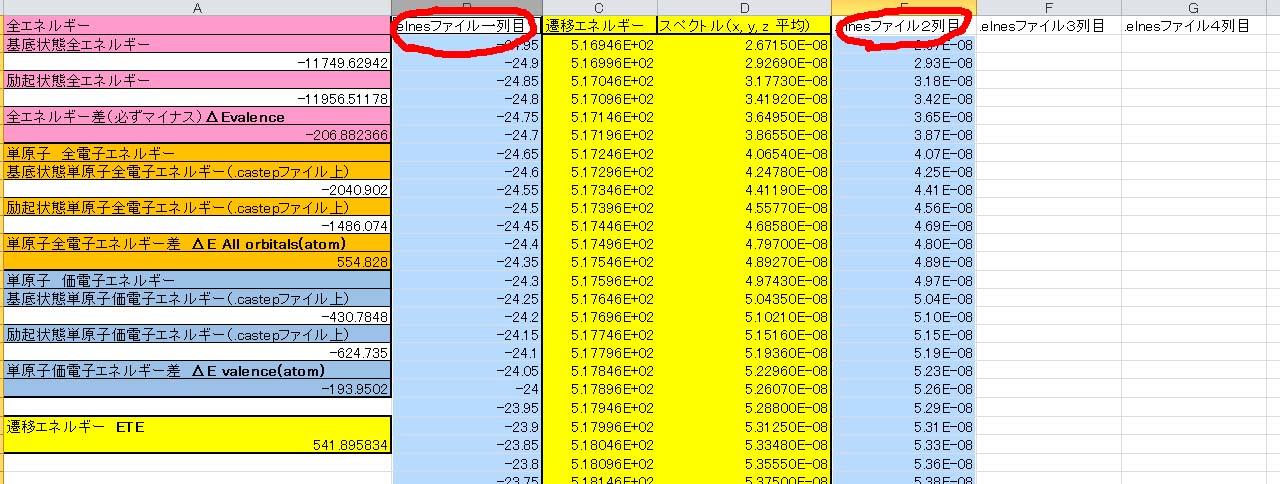
その結果,遷移エネルギーが541.9eVと求められました.この値が計算で得られたELNES/XANESスペクトルの立ち上がりのエネルギーになります.最後に,上で説明した.elnesファイルの目的の部分を別のエクセルファイルに持ってきて,エネルギーとスペクトルを各列にペーストします.
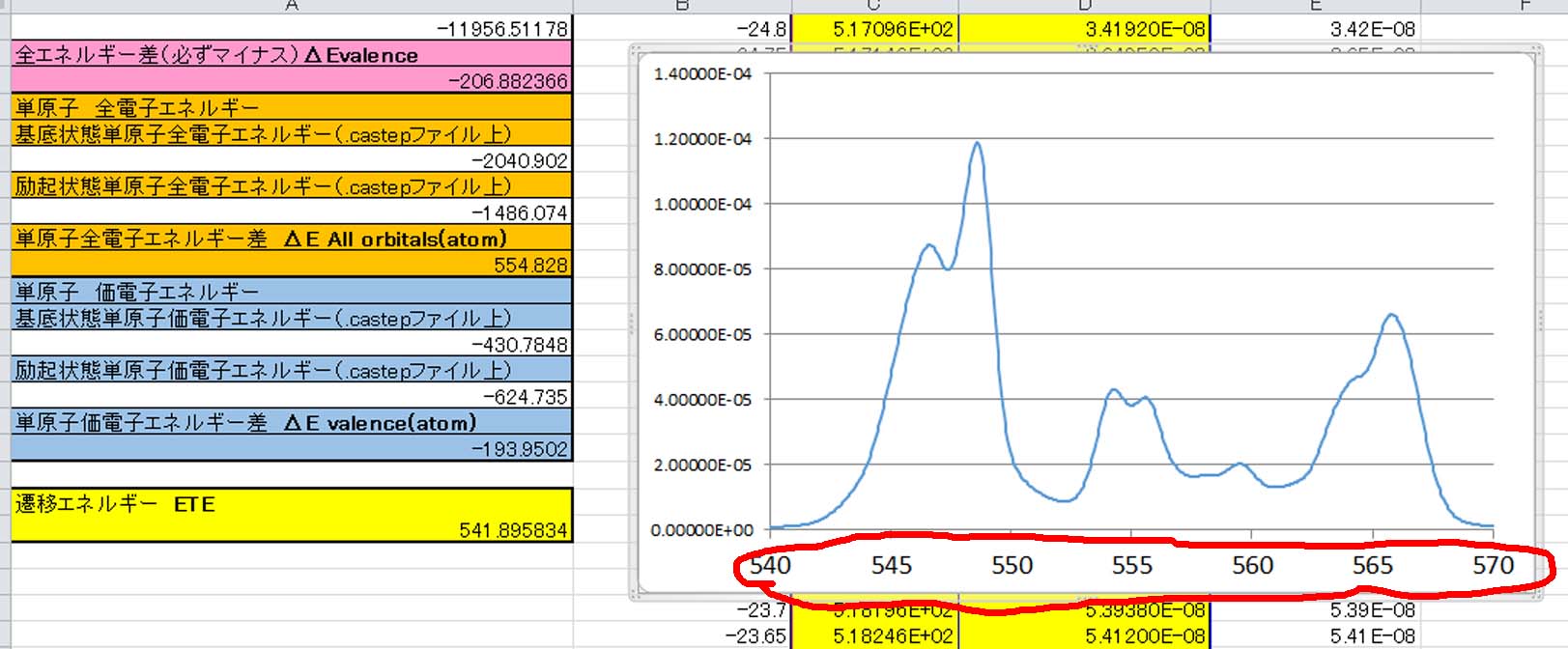
その結果できる真ん中の黄色い2列がエネルギーとスペクトルになります.
スペクトルを書かせると下図のようにエネルギーが求められていることが分かります.

謝辞:ルネサステクノロジー・西藤哲史氏,アクセルリス(BIOVIA)A.
Chatterjee博士,Univ. College London Chris J. Pickard先生,S. P.
Gao先生
補足:CASTEPについて
CASTEPは擬ポテンシャルを用いた第一原理バンド計算コードです.CASTEPについては以下をご参考ください.
S. J.
Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and
M. C. Payne, Z. Kristallogr., 220, 567–570
(2005).
CASTEPを用いたELNES/XANES理論計算については以下をご参照ください.
T. Mizoguchi,
I. Tanaka, S Gao, and C.J. Pickard, J. Phys.: Cond. Matter.,21 (2009)
104204-1-6.